

Description
Xia-Gibbs syndrome is a neurological disorder characterized by weak muscle tone (hypotonia), mild to severe intellectual disability and delayed development. Expressive language skills (vocabulary and the production of speech) are particularly affected; children with this condition usually do not speak their first word, a milestone typically achieved within the first year, until age two or later, and some never learn to talk. Development of motor skills, such as crawling and walking, can also be delayed.
Other signs and symptoms of Xia-Gibbs syndrome vary among affected individuals. Additional neurological features include poor coordination and balance (ataxia) and seizures. Feeding problems and sleep abnormalities can also occur in people with the condition, and many affected individuals experience short pauses in breathing while they sleep (obstructive sleep apnea). In some people with Xia-Gibbs syndrome, imaging tests of the brain show abnormalities in the brain's structure. For example, the tissue connecting the left and right halves of the brain (the corpus callosum) can be abnormally thin.
Xia-Gibbs syndrome can also affect physical development. Growth is usually impaired, and many affected individuals are shorter than their peers. Side-to-side curvature of the spine (scoliosis ) is also a common feature. Some people with Xia-Gibbs syndrome have unusual facial features, such as a broad forehead
) is also a common feature. Some people with Xia-Gibbs syndrome have unusual facial features, such as a broad forehead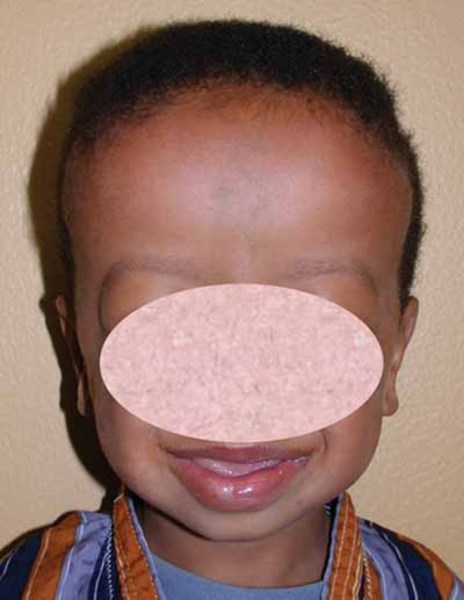 , low-set ears or ears that stick out
, low-set ears or ears that stick out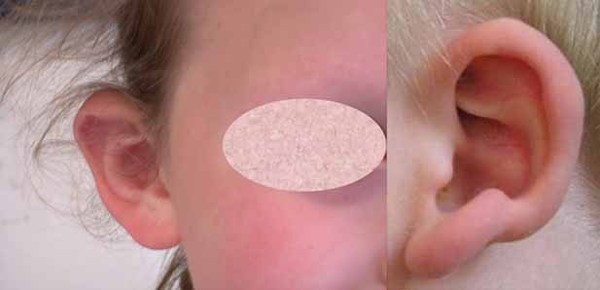 , widely spaced eyes (hypertelorism
, widely spaced eyes (hypertelorism ), eye openings that slant up or down (upslanting palpebral fissures
), eye openings that slant up or down (upslanting palpebral fissures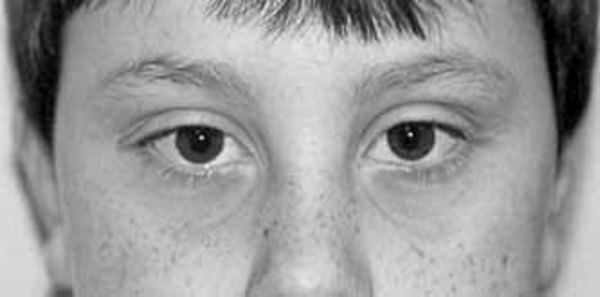 or downslanting palpebral fissures), a flat bridge of the nose, or a thin upper lip
or downslanting palpebral fissures), a flat bridge of the nose, or a thin upper lip . Other, less-common abnormalities involving the bones and skin include premature fusion of certain skull bones (craniosynostosis), unusually loose (lax) joints, and loose skin.
. Other, less-common abnormalities involving the bones and skin include premature fusion of certain skull bones (craniosynostosis), unusually loose (lax) joints, and loose skin.
Neurodevelopmental disorders can also occur in Xia-Gibbs syndrome. Some affected individuals have autism spectrum disorder, which is characterized by impaired communication and social interactions, or attention-deficit/hyperactivity disorder (ADHD). Other problems can include aggression, anxiety, poor impulse control, and self-injury.
Frequency
Xia-Gibbs syndrome is thought to be a rare disorder, although its prevalence is unknown. Doctors believe the condition is underdiagnosed, because many people with intellectual disability never have genetic testing to determine the underlying cause.
Causes
Xia-Gibbs syndrome is caused by variants (also known as mutations) in a gene called AHDC1. This gene provides instructions for making a protein with an unknown function. Researchers suspect that the protein may be able to attach (bind) to DNA and control the activity of other genes. Most of the AHDC1 gene variants involved in Xia-Gibbs syndrome lead to production of abnormally short AHDC1 proteins. The effects of these changes in cells are unclear. The shortened proteins may be quickly broken down or be unable to function. Or, the abnormal proteins may interfere with the function of AHDC1 proteins produced from the normal copy of the gene. Researchers suspect that a reduction in the amount of functional AHDC1 protein impairs normal brain development, leading to intellectual disability, speech problems, and other neurological features of Xia-Gibbs syndrome. Abnormal development of other body systems caused by a shortage of AHDC1 protein may account for additional signs and symptoms of the condition.
Inheritance
Xia-Gibbs syndrome is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
The condition results from new (de novo) variants in the gene that occur either during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. Affected individuals have no history of the disorder in their family.
Other Names for This Condition
- AHDC1-related intellectual disability-obstructive sleep apnea-mild dysmorphism syndrome
- Autosomal dominant intellectual disability 25
- XGS
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chander V, Wangler M, Gibbs R, Murdock D. Xia-Gibbs Syndrome. 2021 Dec 9. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK575793/ Citation on PubMed
- Jiang Y, Wangler MF, McGuire AL, Lupski JR, Posey JE, Khayat MM, Murdock DR, Sanchez-Pulido L, Ponting CP, Xia F, Hunter JV, Meng Q, Murugan M, Gibbs RA. The phenotypic spectrum of Xia-Gibbs syndrome. Am J Med Genet A. 2018 Jun;176(6):1315-1326. doi: 10.1002/ajmg.a.38699. Epub 2018 Apr 25. Citation on PubMed or Free article on PubMed Central
- Murdock DR, Jiang Y, Wangler M, Khayat MM, Sabo A, Juusola J, McWalter K, Schatz KS, Gunay-Aygun M, Gibbs RA. Xia-Gibbs syndrome in adulthood: a case report with insight into the natural history of the condition. Cold Spring Harb Mol Case Stud. 2019 Jun 3;5(3):a003608. doi: 10.1101/mcs.a003608. Print 2019 Jun. Citation on PubMed
- Ritter AL, McDougall C, Skraban C, Medne L, Bedoukian EC, Asher SB, Balciuniene J, Campbell CD, Baker SW, Denenberg EH, Mazzola S, Fiordaliso SK, Krantz ID, Kaplan P, Ierardi-Curto L, Santani AB, Zackai EH, Izumi K. Variable Clinical Manifestations of Xia-Gibbs syndrome: Findings of Consecutively Identified Cases at a Single Children's Hospital. Am J Med Genet A. 2018 Sep;176(9):1890-1896. doi: 10.1002/ajmg.a.40380. Epub 2018 Aug 27. Citation on PubMed
- Wang Q, Huang X, Liu Y, Peng Q, Zhang Y, Liu J, Yuan H. Microdeletion and microduplication of 1p36.11p35.3 involving AHDC1 contribute to neurodevelopmental disorder. Eur J Med Genet. 2020 Jan;63(1):103611. doi: 10.1016/j.ejmg.2019.01.001. Epub 2019 Jan 4. Citation on PubMed
- Xia F, Bainbridge MN, Tan TY, Wangler MF, Scheuerle AE, Zackai EH, Harr MH, Sutton VR, Nalam RL, Zhu W, Nash M, Ryan MM, Yaplito-Lee J, Hunter JV, Deardorff MA, Penney SJ, Beaudet AL, Plon SE, Boerwinkle EA, Lupski JR, Eng CM, Muzny DM, Yang Y, Gibbs RA. De novo truncating mutations in AHDC1 in individuals with syndromic expressive language delay, hypotonia, and sleep apnea. Am J Hum Genet. 2014 May 1;94(5):784-9. doi: 10.1016/j.ajhg.2014.04.006. Citation on PubMed or Free article on PubMed Central
- Yang H, Douglas G, Monaghan KG, Retterer K, Cho MT, Escobar LF, Tucker ME, Stoler J, Rodan LH, Stein D, Marks W, Enns GM, Platt J, Cox R, Wheeler PG, Crain C, Calhoun A, Tryon R, Richard G, Vitazka P, Chung WK. De novo truncating variants in the AHDC1 gene encoding the AT-hook DNA-binding motif-containing protein 1 are associated with intellectual disability and developmental delay. Cold Spring Harb Mol Case Stud. 2015 Oct;1(1):a000562. doi: 10.1101/mcs.a000562. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.