

Description
Von Hippel-Lindau syndrome is an inherited disorder characterized by the formation of tumors and fluid-filled sacs (cysts) in many different parts of the body. Tumors may be either noncancerous or cancerous and most frequently appear during young adulthood; however, the signs and symptoms of von Hippel-Lindau syndrome can occur throughout life.
Tumors called hemangioblastomas are characteristic of von Hippel-Lindau syndrome. These growths are made of newly formed blood vessels. Although they are typically noncancerous, they can cause serious or life-threatening complications. Hemangioblastomas that develop in the brain and spinal cord can cause headaches, vomiting, weakness, and a loss of muscle coordination (ataxia). Hemangioblastomas can also occur in the light-sensitive tissue that lines the back of the eye (the retina). These tumors, which are also called retinal angiomas, may cause vision loss.

People with von Hippel-Lindau syndrome commonly develop cysts in the kidneys, pancreas, and genital tract. They are also at an increased risk of developing a type of kidney cancer called clear cell renal cell carcinoma and a type of pancreatic cancer called a pancreatic neuroendocrine tumor.

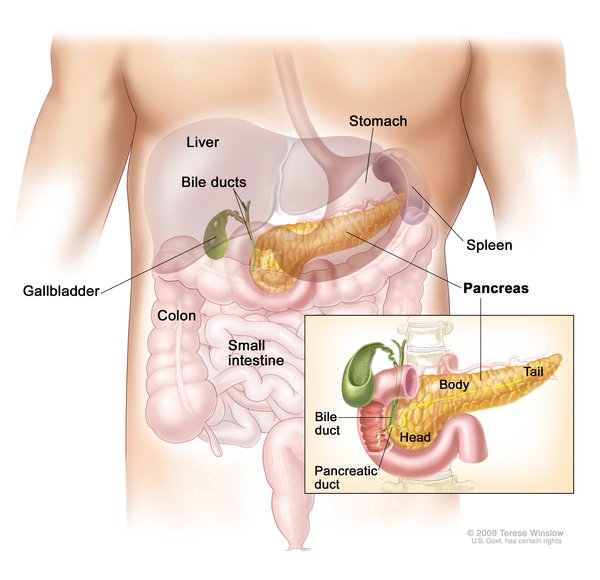
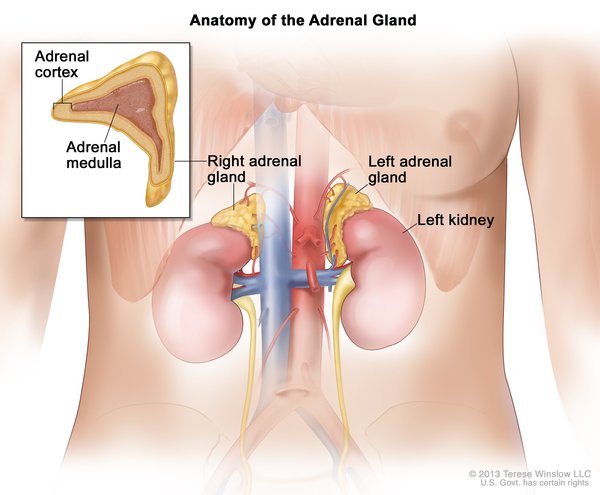
Von Hippel-Lindau syndrome is associated with a type of tumor called a pheochromocytoma, which most commonly occurs in the adrenal glands (small hormone-producing glands located on top of each kidney). Pheochromocytomas are usually noncancerous. They may cause no symptoms, but in some cases they are associated with headaches, panic attacks, excess sweating, or dangerously high blood pressure that may not respond to medication. Pheochromocytomas are particularly dangerous in times of stress or trauma, such as when undergoing surgery or in an accident, or during pregnancy.
About 10 percent of people with von Hippel-Lindau syndrome develop endolymphatic sac tumors, which are noncancerous tumors in the inner ear. These growths can cause hearing loss in one or both ears, as well as ringing in the ears (tinnitus) and problems with balance. Without treatment, these tumors can cause sudden profound deafness.

Noncancerous tumors may also develop in the liver and lungs in people with von Hippel-Lindau syndrome. These tumors do not appear to cause any signs or symptoms.
Frequency
The incidence of von Hippel-Lindau syndrome is estimated to be 1 in 36,000 individuals.
Causes
Mutations in the VHL gene cause von Hippel-Lindau syndrome. The VHL gene is a tumor suppressor gene, which means it keeps cells from growing and dividing too rapidly or in an uncontrolled way. Mutations in this gene prevent production of the VHL protein or lead to the production of an abnormal version of the protein. An altered or missing VHL protein cannot effectively regulate cell survival and division. As a result, cells grow and divide uncontrollably to form the tumors and cysts that are characteristic of von Hippel-Lindau syndrome.
Inheritance
Mutations in the VHL gene are inherited in an autosomal dominant pattern, which means that one copy of the altered gene in each cell is sufficient to increase the risk of developing tumors and cysts. Most people with von Hippel-Lindau syndrome inherit an altered copy of the gene from an affected parent. In about 20 percent of cases, however, the altered gene is the result of a new mutation that occurred during the formation of reproductive cells (eggs or sperm) or very early in development.

Unlike most autosomal dominant conditions, in which one altered copy of a gene in each cell is sufficient to cause the disorder, two copies of the VHL gene must be altered to trigger tumor and cyst formation in von Hippel-Lindau syndrome. A mutation in the second copy of the VHL gene occurs during a person's lifetime in certain cells within organs such as the brain, retina, and kidneys. Cells with two altered copies of this gene do not make functional VHL protein, which allows tumors and cysts to develop. Almost everyone who inherits one VHL mutation will eventually acquire a mutation in the second copy of the gene in some cells, leading to the features of von Hippel-Lindau syndrome.
Other Names for This Condition
- Angiomatosis retinae
- Cerebelloretinal angiomatosis, familial
- Hippel-Lindau disease
- VHL syndrome
- Von Hippel-Lindau disease
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ganeshan D, Menias CO, Pickhardt PJ, Sandrasegaran K, Lubner MG, Ramalingam P, Bhalla S. Tumors in von Hippel-Lindau Syndrome: From Head to Toe-Comprehensive State-of-the-Art Review. Radiographics. 2018 May-Jun;38(3):849-866. doi: 10.1148/rg.2018170156. Epub 2018 Mar 30. Erratum In: Radiographics. 2018 May-Jun;38(3):982. Citation on PubMed
- Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145-73. doi: 10.1146/annurev.pathol.2.010506.092049. Citation on PubMed
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003 Jun 14;361(9374):2059-67. doi: 10.1016/S0140-6736(03)13643-4. Citation on PubMed
- Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011 Jun;19(6):617-23. doi: 10.1038/ejhg.2010.175. Epub 2011 Mar 9. Citation on PubMed or Free article on PubMed Central
- Richard S, Graff J, Lindau J, Resche F. Von Hippel-Lindau disease. Lancet. 2004 Apr 10;363(9416):1231-4. doi: 10.1016/S0140-6736(04)15957-6. No abstract available. Citation on PubMed
- Sano T, Horiguchi H. Von Hippel-Lindau disease. Microsc Res Tech. 2003 Feb 1;60(2):159-64. doi: 10.1002/jemt.10253. Citation on PubMed
- Shehata BM, Stockwell CA, Castellano-Sanchez AA, Setzer S, Schmotzer CL, Robinson H. Von Hippel-Lindau (VHL) disease: an update on the clinico-pathologic and genetic aspects. Adv Anat Pathol. 2008 May;15(3):165-71. doi: 10.1097/PAP.0b013e31816f852e. Citation on PubMed
- Shuin T, Yamasaki I, Tamura K, Okuda H, Furihata M, Ashida S. Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol. 2006 Jun;36(6):337-43. doi: 10.1093/jjco/hyl052. Citation on PubMed
- van Leeuwaarde RS, Ahmad S, van Nesselrooij B, Zandee W, Giles RH. Von Hippel-Lindau Syndrome. 2000 May 17 [updated 2024 Feb 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1463/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.