

Description
TUBB4A-related leukodystrophy is a disorder that affects the nervous system. Leukodystrophies are conditions that involve abnormalities of the nervous system's white matter, which consists of nerve fibers covered by a fatty substance called myelin. Myelin insulates nerve fibers and promotes the rapid transmission of nerve impulses. In particular, TUBB4A-related leukodystrophy involves hypomyelination, which means that the nervous system has a reduced ability to form myelin. In some affected individuals, myelin may also break down, which is known as demyelination.

People with TUBB4A-related leukodystrophy have different combinations of signs and symptoms. Some of these combinations are described as separate disorders. However, the features in some affected individuals do not fit into these defined disorders. Researchers now group all of these cases of leukodystrophy, which have the same genetic cause, as TUBB4A-related leukodystrophy.
At the most severe end of the TUBB4A-related leukodystrophy spectrum is a condition called hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). This disorder begins in infancy or early childhood. Most affected individuals have delayed development of motor skills, such as sitting and walking, and some are never able to walk on their own. In other cases, motor skills develop normally and then are lost in early childhood (developmental regression). In addition, individuals with H-ABC have other movement abnormalities, such as involuntary muscle contractions (dystonia), uncontrolled movements of the limbs (choreoathetosis), muscle stiffness (rigidity), and difficulty coordinating movements (ataxia). These individuals also often have impaired speech (dysarthria), a weak voice (dysphonia), and swallowing problems (dysphagia). Some develop seizures. Learning difficulty is common in individuals with H-ABC.
H-ABC is characterized by particular brain abnormalities, including hypomyelination. In addition, tissue in certain regions of the brain breaks down (atrophies), most prominently in a region called the putamen, which is part of a group of structures that help control movement (the basal ganglia). Atrophy of brain tissue in another region involved in movement called the cerebellum is common, and atrophy of the cerebrum, which controls most voluntary activity, language, sensory perception, learning, and memory, can also occur.
At the mildest end of the TUBB4A-related leukodystrophy spectrum is a condition called isolated hypomyelination, which begins at any time from late childhood to adulthood. Individuals at this end of the spectrum have mild hypomyelination and sometimes mild atrophy of the cerebellum, but no problems with the basal ganglia. These individuals can have movement problems, dysarthria, and learning difficulty, although these features are typically milder than in H-ABC.
The features in other individuals with TUBB4A-related leukodystrophy fall in between these two extremes. Affected individuals can have varying degrees of hypomyelination and atrophy or impairment of the basal ganglia or other brain regions. Movement problems can also occur. A small group of affected individuals develop muscle stiffness and paralysis of the lower limbs (spastic paraplegia) that slowly worsen. In addition, these individuals may have mild hypomyelination and ataxia without the other movement or learning problems common in H-ABC.
Frequency
TUBB4A-related leukodystrophy is a rare disorder, although the exact prevalence of the condition is unknown. At least 70 affected individuals have been described in the medical literature.
Causes
TUBB4A-related leukodystrophy is caused by mutations in the TUBB4A gene, which provides instructions for making a protein called beta-tubulin (β-tubulin). This protein attaches to another protein called alpha-tubulin (α-tubulin) to form structures called microtubules, which form the framework of cells (cytoskeleton). β-tubulin produced from the TUBB4A gene is found primarily in the brain, particularly in the putamen, cerebellum, and white matter. During brain development, microtubules help move nerve cells (neurons) to their proper location (neuronal migration). The microtubules also form scaffolding within neurons that provides structure and aids in the transport of substances.

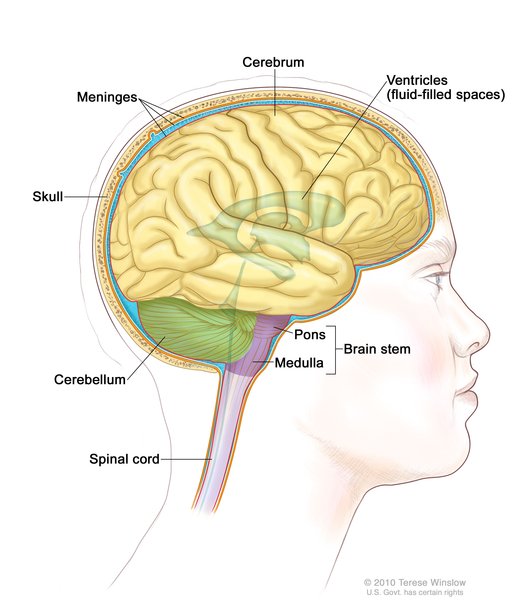
The mutations that cause TUBB4A-related leukodystrophy are thought to alter the structure of the β-tubulin protein, likely impairing the formation or stability of microtubules. While it is unclear how these genetic changes lead to the signs and symptoms of TUBB4A-related leukodystrophy, researchers suspect that problems with microtubules impair neuronal migration or the transport of important substances within neurons, which may lead to dysfunction and loss of these cells in the brain, particularly in the putamen, cerebellum, and white matter. Abnormalities in these brain regions underlie the movement, speech, and learning problems that can occur in TUBB4A-related leukodystrophy. Researchers do not understand what causes the wide range of severity in this disorder.
Inheritance
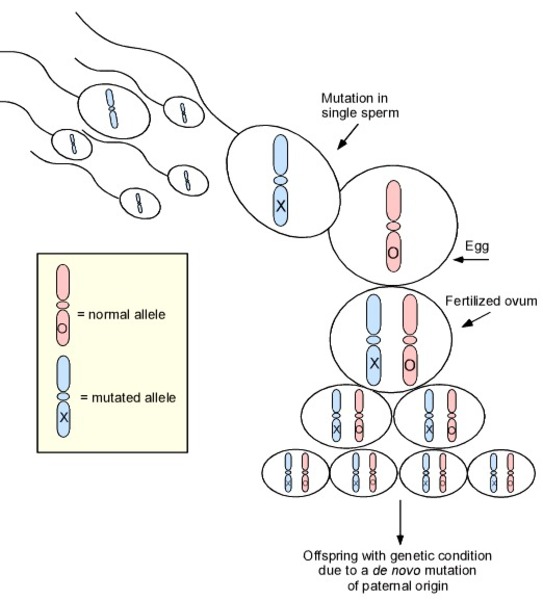
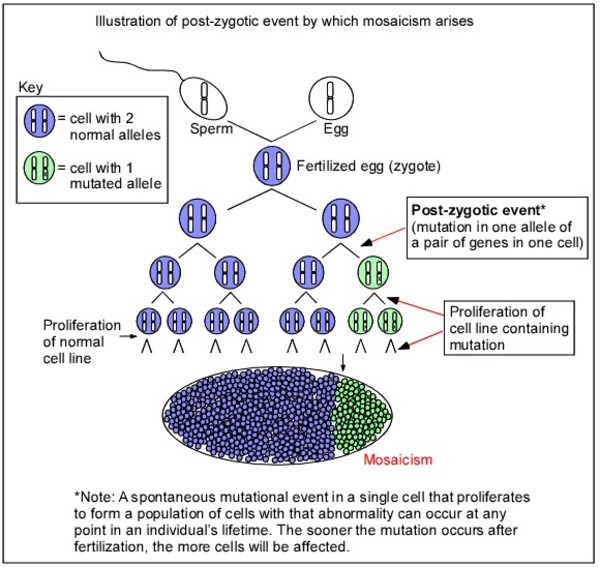
TUBB4A-related leukodystrophy is inherited in an autosomal dominant pattern, which means one copy of the altered TUBB4A gene in each cell is sufficient to cause the disorder. Most cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the disorder in their family. Very rarely, the condition is inherited from a parent with mosaicism. Mosaicism means that some of the body's cells have the gene mutation, and others do not. In these instances, the parent with mosaicism does not show any signs or symptoms of TUBB4A-related leukodystrophy.

Other Names for This Condition
- TUBB4A-associated hypomyelinating leukoencephalopathies
- TUBB4A-related hypomyelinating leukodystrophy
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Blumkin L, Halevy A, Ben-Ami-Raichman D, Dahari D, Haviv A, Sarit C, Lev D, van der Knaap MS, Lerman-Sagie T, Leshinsky-Silver E. Expansion of the spectrum of TUBB4A-related disorders: a new phenotype associated with a novel mutation in the TUBB4A gene. Neurogenetics. 2014 May;15(2):107-13. doi: 10.1007/s10048-014-0392-2. Epub 2014 Feb 14. Erratum In: Neurogenetics. 2014 May; 15(2):115. Citation on PubMed
- Hamilton EM, Polder E, Vanderver A, Naidu S, Schiffmann R, Fisher K, Raguz AB, Blumkin L; H-ABC Research Group; van Berkel CG, Waisfisz Q, Simons C, Taft RJ, Abbink TE, Wolf NI, van der Knaap MS. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain. 2014 Jul;137(Pt 7):1921-30. doi: 10.1093/brain/awu110. Epub 2014 Apr 30. Citation on PubMed or Free article on PubMed Central
- Kancheva D, Chamova T, Guergueltcheva V, Mitev V, Azmanov DN, Kalaydjieva L, Tournev I, Jordanova A. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia. Mov Disord. 2015 May;30(6):854-8. doi: 10.1002/mds.26196. Epub 2015 Mar 15. Citation on PubMed
- Miyatake S, Osaka H, Shiina M, Sasaki M, Takanashi J, Haginoya K, Wada T, Morimoto M, Ando N, Ikuta Y, Nakashima M, Tsurusaki Y, Miyake N, Ogata K, Matsumoto N, Saitsu H. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology. 2014 Jun 17;82(24):2230-7. doi: 10.1212/WNL.0000000000000535. Epub 2014 May 21. Citation on PubMed
- Nahhas N, Conant A, Hamilton E, Curiel J, Simons C, van der Knaap M, Vanderver A. TUBB4A-Related Leukodystrophy. 2016 Nov 3. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK395611/ Citation on PubMed
- Pizzino A, Pierson TM, Guo Y, Helman G, Fortini S, Guerrero K, Saitta S, Murphy JL, Padiath Q, Xie Y, Hakonarson H, Xu X, Funari T, Fox M, Taft RJ, van der Knaap MS, Bernard G, Schiffmann R, Simons C, Vanderver A. TUBB4A de novo mutations cause isolated hypomyelination. Neurology. 2014 Sep 2;83(10):898-902. doi: 10.1212/WNL.0000000000000754. Epub 2014 Aug 1. Citation on PubMed or Free article on PubMed Central
- Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A, Crawford J, Ru K, Grimmond SM, Miller D, Tonduti D, Schmidt JL, Chudnow RS, van Coster R, Lagae L, Kisler J, Sperner J, van der Knaap MS, Schiffmann R, Taft RJ, Vanderver A. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet. 2013 May 2;92(5):767-73. doi: 10.1016/j.ajhg.2013.03.018. Epub 2013 Apr 11. Citation on PubMed or Free article on PubMed Central
- Tonduti D, Aiello C, Renaldo F, Dorboz I, Saaman S, Rodriguez D, Fettah H, Elmaleh M, Biancheri R, Barresi S, Boccone L, Orcesi S, Pichiecchio A, Zangaglia R, Maurey H, Rossi A, Boespflug-Tanguy O, Bertini E. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur J Paediatr Neurol. 2016 Mar;20(2):323-330. doi: 10.1016/j.ejpn.2015.11.006. Epub 2015 Nov 28. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.