

Description
Thrombocytopenia-absent radius (TAR) syndrome is characterized by the absence of a bone called the radius in each forearm and a shortage (deficiency) of blood cells involved in clotting (platelets). This platelet deficiency (thrombocytopenia) usually appears during infancy and becomes less severe over time; in some cases the platelet levels become normal.
Thrombocytopenia prevents normal blood clotting, resulting in easy bruising and frequent nosebleeds. Potentially life-threatening episodes of severe bleeding (hemorrhages) may occur in the brain and other organs, especially during the first year of life. Hemorrhages can damage the brain and lead to intellectual disability. Affected children who survive this period and do not have damaging hemorrhages in the brain usually have a normal life expectancy and normal intellectual development.
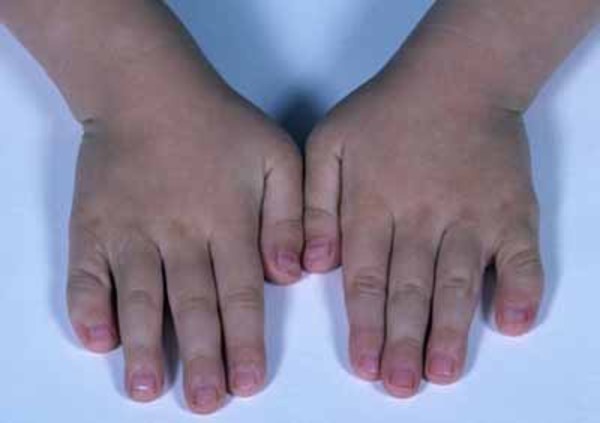
The severity of skeletal problems in TAR syndrome varies among affected individuals. The radius, which is the bone on the thumb side of the forearm, is almost always missing in both arms. The other bone in the forearm, which is called the ulna, is sometimes underdeveloped or absent in one or both arms. TAR syndrome is unusual among similar malformations in that affected individuals have thumbs, while people with other conditions involving an absent radius typically do not. However, there may be other abnormalities of the hands, such as webbed or fused fingers (syndactyly) or curved pinky fingers (fifth finger clinodactyly). Some people with TAR syndrome also have skeletal abnormalities affecting the upper arms, legs, or hip sockets.
Other features that can occur in TAR syndrome include malformations of the heart or kidneys. Some people with this disorder have unusual facial features including a small lower jaw (micrognathia), a prominent forehead, and low-set ears. About half of affected individuals have allergic reactions to cow's milk that may worsen the thrombocytopenia associated with this disorder.
Frequency
TAR syndrome is a rare disorder, affecting fewer than 1 in 100,000 newborns.
Causes
Mutations in the RBM8A gene cause TAR syndrome. The RBM8A gene provides instructions for making a protein called RNA-binding motif protein 8A. This protein is believed to be involved in several important cellular functions involving the production of other proteins.
Most people with TAR syndrome have a mutation in one copy of the RBM8A gene and a deletion of genetic material from chromosome 1 that includes the other copy of the RBM8A gene in each cell. A small number of affected individuals have mutations in both copies of the RBM8A gene in each cell and do not have a deletion on chromosome 1. RBM8A gene mutations that cause TAR syndrome reduce the amount of RNA-binding motif protein 8A in cells. The deletions involved in TAR syndrome eliminate at least 200,000 DNA building blocks (200 kilobases, or 200 kb) from the long (q) arm of chromosome 1 in a region called 1q21.1. The deletion eliminates one copy of the RBM8A gene in each cell and the RNA-binding motif protein 8A that would have been produced from it.
People with either an RBM8A gene mutation and a chromosome 1 deletion or with two gene mutations have a decreased amount of RNA-binding motif protein 8A. This reduction is thought to cause problems in the development of certain tissues, but it is unknown how it causes the specific signs and symptoms of TAR syndrome. No cases have been reported in which a deletion that includes the RBM8A gene occurs on both copies of chromosome 1; studies indicate that the complete loss of RNA-binding motif protein 8A is not compatible with life.
Researchers sometimes refer to the deletion in chromosome 1 associated with TAR syndrome as the 200-kb deletion to distinguish it from another chromosomal abnormality called a 1q21.1 microdeletion. People with a 1q21.1 microdeletion are missing a different, larger DNA segment in the chromosome 1q21.1 region near the area where the 200-kb deletion occurs. The chromosomal change related to 1q21.1 microdeletion is often called the recurrent distal 1.35-Mb deletion.
Inheritance
TAR syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell are altered. In this disorder, either both copies of the RBM8A gene in each cell have mutations or, more commonly, one copy of the gene has a mutation and the other is lost as part of a deleted segment on chromosome 1. The affected individual usually inherits an RBM8A gene mutation from one parent. In about 75 percent of cases, the affected person inherits a copy of chromosome 1 with the 200-kb deletion from the other parent. In the remaining cases, the deletion occurs during the formation of reproductive cells (eggs and sperm) or in early fetal development. Although parents of an individual with TAR syndrome can carry an RBM8A gene mutation or a 200-kb deletion, they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Chromosome 1q21.1 deletion syndrome, 200-KB
- Radial aplasia-amegakaryocytic thrombocytopenia
- Radial aplasia-thrombocytopenia syndrome
- TAR syndrome
- Thrombocytopenia absent radii
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, Breuning MH, Debili N, Deloukas P, Favier R, Fiedler J, Hobbs CM, Huang N, Hurles ME, Kiddle G, Krapels I, Nurden P, Ruivenkamp CA, Sambrook JG, Smith K, Stemple DL, Strauss G, Thys C, van Geet C, Newbury-Ecob R, Ouwehand WH, Ghevaert C. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012 Feb 26;44(4):435-9, S1-2. doi: 10.1038/ng.1083. Citation on PubMed or Free article on PubMed Central
- Bonsi L, Marchionni C, Alviano F, Lanzoni G, Franchina M, Costa R, Grossi A, Bagnara GP. Thrombocytopenia with absent radii (TAR) syndrome: from hemopoietic progenitor to mesenchymal stromal cell disease? Exp Hematol. 2009 Jan;37(1):1-7. doi: 10.1016/j.exphem.2008.09.004. Epub 2008 Nov 22. Citation on PubMed
- de Ybarrondo L, Barratt MS. Thrombocytopenia absent radius syndrome. Pediatr Rev. 2011 Sep;32(9):399-400; discussion 400. doi: 10.1542/pir.32-9-399. No abstract available. Citation on PubMed
- Greenhalgh KL, Howell RT, Bottani A, Ancliff PJ, Brunner HG, Verschuuren-Bemelmans CC, Vernon E, Brown KW, Newbury-Ecob RA. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet. 2002 Dec;39(12):876-81. doi: 10.1136/jmg.39.12.876. Citation on PubMed or Free article on PubMed Central
- Houeijeh A, Andrieux J, Saugier-Veber P, David A, Goldenberg A, Bonneau D, Fouassier M, Journel H, Martinovic J, Escande F, Devisme L, Bisiaux S, Chaffiotte C, Baux M, Kerckaert JP, Holder-Espinasse M, Manouvrier-Hanu S. Thrombocytopenia-absent radius (TAR) syndrome: a clinical genetic series of 14 further cases. impact of the associated 1q21.1 deletion on the genetic counselling. Eur J Med Genet. 2011 Sep-Oct;54(5):e471-7. doi: 10.1016/j.ejmg.2011.05.001. Epub 2011 May 13. Citation on PubMed
- Klopocki E, Schulze H, Strauss G, Ott CE, Hall J, Trotier F, Fleischhauer S, Greenhalgh L, Newbury-Ecob RA, Neumann LM, Habenicht R, Konig R, Seemanova E, Megarbane A, Ropers HH, Ullmann R, Horn D, Mundlos S. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007 Feb;80(2):232-40. doi: 10.1086/510919. Epub 2006 Dec 21. Citation on PubMed or Free article on PubMed Central
- Mokha J, Serrano M. Thrombocytopenia associated with cow's milk protein allergy: a case report. Clin Pediatr (Phila). 2013 Oct;52(10):985-7. doi: 10.1177/0009922812456593. Epub 2012 Aug 20. No abstract available. Citation on PubMed
- Toriello HV. Thrombocytopenia-absent radius syndrome. Semin Thromb Hemost. 2011 Sep;37(6):707-12. doi: 10.1055/s-0031-1291381. Epub 2011 Nov 18. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.