

Description
Succinate-CoA ligase deficiency is an inherited disorder that affects the early development of the brain and other body systems. One of the earliest signs of the disorder is very weak muscle tone (severe hypotonia), which appears in the first few months of life. Severe hypotonia delays the development of motor skills such as holding up the head and rolling over. Many affected children also have muscle weakness and reduced muscle mass, which prevents them from standing and walking independently.
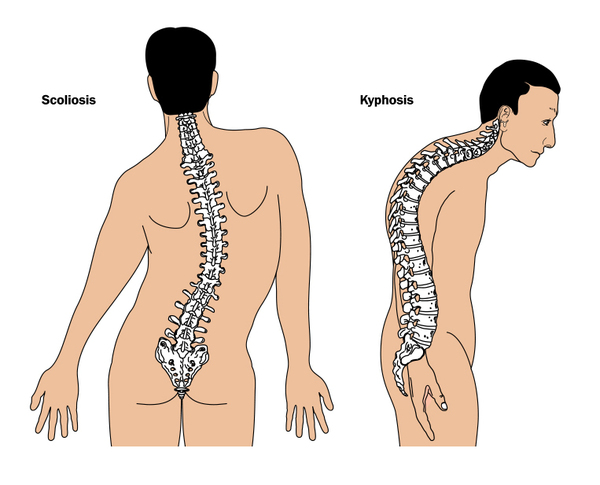
Additional features of succinate-CoA ligase deficiency can include progressive abnormal curvature of the spine (scoliosis or kyphosis), uncontrolled movements (dystonia), severe hearing loss, and seizures beginning in childhood. In most affected children, a substance called methylmalonic acid builds up abnormally in the body and is excreted in urine (methylmalonic aciduria). Most children with succinate-CoA ligase deficiency also experience a failure to thrive, which means that they gain weight and grow more slowly than expected.
Succinate-CoA ligase deficiency causes breathing difficulties that often lead to recurrent infections of the respiratory tract. These infections can be life-threatening, and most people with succinate-CoA ligase deficiency live only into childhood or adolescence.
A few individuals with succinate-CoA ligase deficiency have had an even more severe form of the disorder known as fatal infantile lactic acidosis. Affected infants develop a toxic buildup of lactic acid in the body (lactic acidosis) in the first day of life, which leads to muscle weakness and breathing difficulties. Children with fatal infantile lactic acidosis usually live only a few days after birth.
Frequency
Although the exact prevalence of succinate-CoA ligase deficiency is unknown, it appears to be very rare. This condition occurs more frequently among people from the Faroe Islands in the North Atlantic Ocean.
Causes
Succinate-CoA ligase deficiency results from mutations in the SUCLA2 or SUCLG1 gene. SUCLG1 gene mutations can cause fatal infantile lactic acidosis, while mutations in either gene can cause the somewhat less severe form of the condition.
The SUCLA2 and SUCLG1 genes each provide instructions for making one part (subunit) of an enzyme called succinate-CoA ligase. This enzyme plays a critical role in mitochondria, which are structures within cells that convert the energy from food into a form that cells can use. Mitochondria each contain a small amount of DNA, known as mitochondrial DNA or mtDNA, which is essential for the normal function of these structures. Succinate-CoA ligase is involved in producing and maintaining the building blocks of mitochondrial DNA.
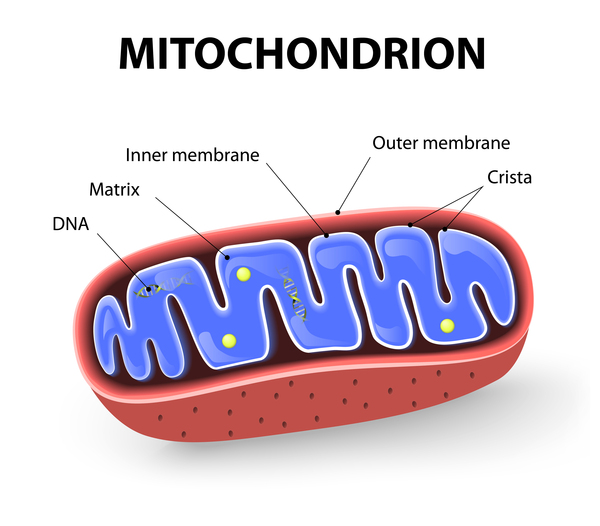
Mutations in either the SUCLA2 or SUCLG1 gene disrupt the normal function of succinate-CoA ligase. A shortage (deficiency) of this enzyme leads to problems with the production and maintenance of mitochondrial DNA. A reduction in the amount of mitochondrial DNA (known as mitochondrial DNA depletion) impairs mitochondrial function in many of the body's cells and tissues. These problems lead to hypotonia, muscle weakness, and the other characteristic features of succinate-CoA ligase deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Mitochondrial DNA depletion syndrome, encephalomyopathic form, with mild methylmalonic aciduria
- Mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria
- Succinate-coenzyme A ligase deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Scientific Articles on PubMed
References
- Alberio S, Mineri R, Tiranti V, Zeviani M. Depletion of mtDNA: syndromes and genes. Mitochondrion. 2007 Feb-Apr;7(1-2):6-12. doi: 10.1016/j.mito.2006.11.010. Epub 2006 Dec 5. Citation on PubMed
- Carrozzo R, Dionisi-Vici C, Steuerwald U, Lucioli S, Deodato F, Di Giandomenico S, Bertini E, Franke B, Kluijtmans LA, Meschini MC, Rizzo C, Piemonte F, Rodenburg R, Santer R, Santorelli FM, van Rooij A, Vermunt-de Koning D, Morava E, Wevers RA. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007 Mar;130(Pt 3):862-74. doi: 10.1093/brain/awl389. Epub 2007 Feb 14. Citation on PubMed
- Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005 Jun;76(6):1081-6. doi: 10.1086/430843. Epub 2005 Apr 22. Citation on PubMed or Free article on PubMed Central
- Ostergaard E, Christensen E, Kristensen E, Mogensen B, Duno M, Shoubridge EA, Wibrand F. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007 Aug;81(2):383-7. doi: 10.1086/519222. Epub 2007 Jun 4. Citation on PubMed or Free article on PubMed Central
- Ostergaard E, Hansen FJ, Sorensen N, Duno M, Vissing J, Larsen PL, Faeroe O, Thorgrimsson S, Wibrand F, Christensen E, Schwartz M. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007 Mar;130(Pt 3):853-61. doi: 10.1093/brain/awl383. Epub 2007 Feb 7. Citation on PubMed
- Ostergaard E, Schwartz M, Batbayli M, Christensen E, Hjalmarson O, Kollberg G, Holme E. A novel missense mutation in SUCLG1 associated with mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria. Eur J Pediatr. 2010 Feb;169(2):201-5. doi: 10.1007/s00431-009-1007-z. Epub 2009 Jun 14. Citation on PubMed
- Ostergaard E. Disorders caused by deficiency of succinate-CoA ligase. J Inherit Metab Dis. 2008 Apr;31(2):226-9. doi: 10.1007/s10545-008-0828-7. Epub 2008 Apr 4. Citation on PubMed
- Spinazzola A, Invernizzi F, Carrara F, Lamantea E, Donati A, Dirocco M, Giordano I, Meznaric-Petrusa M, Baruffini E, Ferrero I, Zeviani M. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. 2009 Apr;32(2):143-58. doi: 10.1007/s10545-008-1038-z. Epub 2008 Dec 27. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.