

Description
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) is a severe skin reaction most often triggered by particular medications. Although Stevens-Johnson syndrome and toxic epidermal necrolysis were once thought to be separate conditions, they are now considered part of a continuum. Stevens-Johnson syndrome represents the less severe end of the disease spectrum, and toxic epidermal necrolysis represents the more severe end.
SJS/TEN often begins with a fever and flu-like symptoms. Within a few days, the skin begins to blister and peel, forming very painful raw areas called erosions that resemble a severe hot-water burn. The skin erosions usually start on the face and chest before spreading to other parts of the body. In most affected individuals, the condition also damages the mucous membranes, including the lining of the mouth and the airways, which can cause trouble with swallowing and breathing. The painful blistering can also affect the urinary tract and genitals. SJS/TEN often affects the eyes as well, causing irritation and redness of the conjunctiva, which are the mucous membranes that protect the white part of the eye and line the eyelids, and damage to the clear front covering of the eye (the cornea).

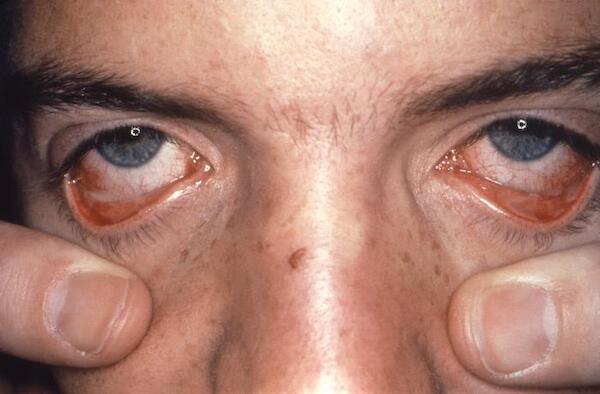

Severe damage to the skin and mucous membranes makes SJS/TEN a life-threatening disease. Because the skin normally acts as a protective barrier, extensive skin damage can lead to a dangerous loss of fluids and allow infections to develop. Serious complications can include pneumonia, overwhelming bacterial infections (sepsis), shock, multiple organ failure, and death. About 10 percent of people with Stevens-Johnson syndrome die from the disease, while the condition is fatal in up to 50 percent of those with toxic epidermal necrolysis.
Among people who survive, long-term effects of SJS/TEN can include changes in skin coloring (pigmentation), dryness of the skin and mucous membranes (xerosis), excess sweating (hyperhidrosis), hair loss (alopecia), and abnormal growth or loss of the fingernails and toenails. Other long-term problems can include impaired taste, difficulty urinating, and genital abnormalities. A small percentage of affected individuals develop chronic dryness or inflammation of the eyes, which can lead to increased sensitivity to light (photophobia) and vision impairment.
Frequency
SJS/TEN is a rare disease, affecting 1 to 2 per million people each year. Stevens-Johnson syndrome (the less severe form of the condition) is more common than toxic epidermal necrolysis.
People who are HIV-positive and those with a chronic inflammatory disease called systemic lupus erythematosus are more likely to develop SJS/TEN than the general population. The reason for the increased risk is unclear, but immune system factors and exposure to multiple medications may play a role.
Causes
Several genetic changes have been found to increase the risk of SJS/TEN in response to triggering factors such as medications. Most of these changes occur in genes that are involved in the normal function of the immune system.
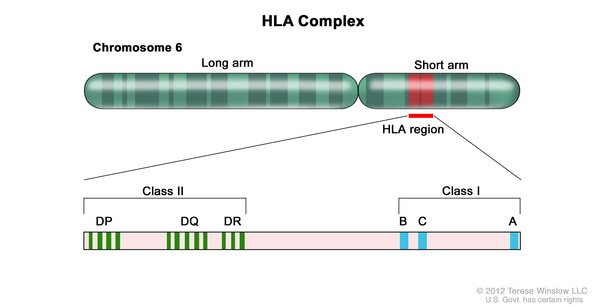
The genetic variations most strongly associated with SJS/TEN occur in the HLA-B gene. This gene is part of a family of genes called the human leukocyte antigen (HLA) complex. The HLA complex helps the immune system distinguish the body's own proteins from proteins made by foreign invaders (such as viruses and bacteria). The HLA-B gene has many different normal variations, allowing each person's immune system to react to a wide range of foreign proteins. Certain variations in this gene occur much more often in people with SJS/TEN than in people without the condition.
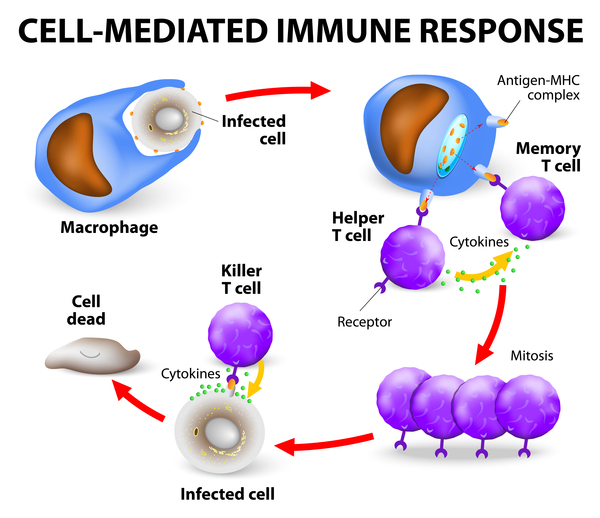
Studies suggest that the HLA-B gene variations associated with SJS/TEN cause the immune system to react abnormally to certain medications. In a process that is not well understood, the drug causes immune cells called cytotoxic T cells and natural killer (NK) cells to release a substance called granulysin that destroys cells in the skin and mucous membranes. The death of these cells causes the blistering and peeling that is characteristic of SJS/TEN.
Variations in several other HLA and non-HLA genes have also been studied as potential risk factors for SJS/TEN. However, most people with genetic variations that increase the risk of SJS/TEN never develop the disease, even if they are exposed to drugs that can trigger it. Researchers believe that additional genetic and nongenetic factors, many of which are unknown, likely play a role in whether a particular individual develops SJS/TEN.
The drugs most frequently associated with SJS/TEN include several medications that are used to treat seizures (particularly carbamazepine, lamotrigine, and phenytoin); allopurinol, which is used to treat kidney stones and a form of arthritis called gout; a class of antibiotic drugs called sulfonamides; nevirapine, which is used to treat HIV infection; and a type of non-steroidal anti-inflammatory drugs (NSAIDs) called oxicams. Other factors may also trigger SJS/TEN. In particular, these skin reactions have occurred in people with an unusual form of pneumonia caused by infection with Mycoplasma pneumoniae and in people with viral infections, including cytomegalovirus. Researchers suspect that a combination of infections and drugs could contribute to the disease in some individuals. In many cases, no definitive trigger for an individual's SJS/TEN is ever discovered.
Inheritance
SJS/TEN is not an inherited condition. However, the genetic changes that increase the risk of developing SJS/TEN can be passed from one generation to the next.
Other Names for This Condition
- Drug-induced Stevens Johnson syndrome
- Lyell's syndrome
- Mycoplasma-induced Stevens Johnson syndrome
- Stevens-Johnson syndrome
- Stevens-Johnson syndrome toxic epidermal necrolysis spectrum
- Toxic epidermal necrolysis
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Amstutz U, Shear NH, Rieder MJ, Hwang S, Fung V, Nakamura H, Connolly MB, Ito S, Carleton BC; CPNDS clinical recommendation group. Recommendations for HLA-B*15:02 and HLA-A*31:01 genetic testing to reduce the risk of carbamazepine-induced hypersensitivity reactions. Epilepsia. 2014 Apr;55(4):496-506. doi: 10.1111/epi.12564. Epub 2014 Mar 5. Citation on PubMed
- Cheng CY, Su SC, Chen CH, Chen WL, Deng ST, Chung WH. HLA associations and clinical implications in T-cell mediated drug hypersensitivity reactions: an updated review. J Immunol Res. 2014;2014:565320. doi: 10.1155/2014/565320. Epub 2014 May 8. Citation on PubMed or Free article on PubMed Central
- Chung WH, Hung SI, Yang JY, Su SC, Huang SP, Wei CY, Chin SW, Chiou CC, Chu SC, Ho HC, Yang CH, Lu CF, Wu JY, Liao YD, Chen YT. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med. 2008 Dec;14(12):1343-50. doi: 10.1038/nm.1884. Epub 2008 Nov 23. Citation on PubMed
- Chung WH, Hung SI. Recent advances in the genetics and immunology of Stevens-Johnson syndrome and toxic epidermal necrosis. J Dermatol Sci. 2012 Jun;66(3):190-6. doi: 10.1016/j.jdermsci.2012.04.002. Epub 2012 Apr 11. Citation on PubMed
- Lee HY, Chung WH. Toxic epidermal necrolysis: the year in review. Curr Opin Allergy Clin Immunol. 2013 Aug;13(4):330-6. doi: 10.1097/ACI.0b013e3283630cc2. Citation on PubMed
- Mockenhaupt M. Stevens-Johnson syndrome and toxic epidermal necrolysis: clinical patterns, diagnostic considerations, etiology, and therapeutic management. Semin Cutan Med Surg. 2014 Mar;33(1):10-6. doi: 10.12788/j.sder.0058. Citation on PubMed
- Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, Oseroff C, Lu S, Jakoncic J, de Oliveira CA, Yang L, Mei H, Shi L, Shabanowitz J, English AM, Wriston A, Lucas A, Phillips E, Mallal S, Grey HM, Sette A, Hunt DF, Buus S, Peters B. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci U S A. 2012 Jun 19;109(25):9959-64. doi: 10.1073/pnas.1207934109. Epub 2012 May 29. Citation on PubMed or Free article on PubMed Central
- Sukasem C, Katsila T, Tempark T, Patrinos GP, Chantratita W. Drug-Induced Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis Call for Optimum Patient Stratification and Theranostics via Pharmacogenomics. Annu Rev Genomics Hum Genet. 2018 Aug 31;19:329-353. doi: 10.1146/annurev-genom-083115-022324. Epub 2018 Apr 13. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.