

Description
Spinocerebellar ataxia type 36 (SCA36) is a condition characterized by progressive problems with movement that typically begin in mid-adulthood. People with this condition initially experience problems with coordination and balance (ataxia). Affected individuals often have exaggerated reflexes (hyperreflexia) and problems with speech (dysarthria). They also usually develop muscle twitches (fasciculations) of the tongue and over time, the muscles in the tongue waste away (atrophy). These tongue problems can cause difficulties swallowing liquids. As the condition progresses, individuals with SCA36 develop muscle atrophy in the legs, forearms, and hands. Another common feature of SCA36 is the atrophy of specialized nerve cells that control muscle movement (motor neurons), which can contribute to the tongue and limb muscle atrophy in affected individuals.
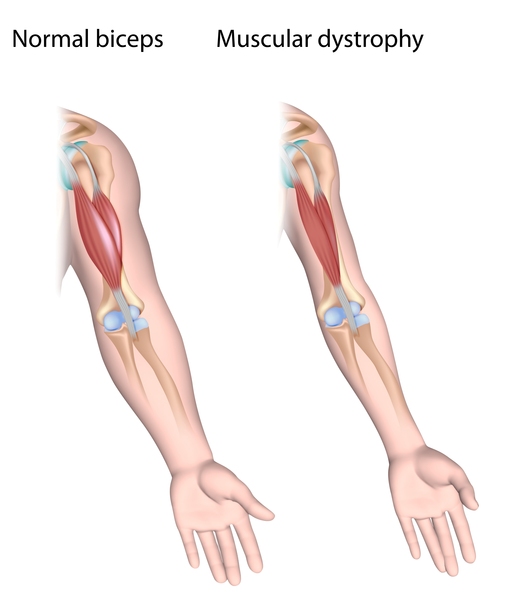
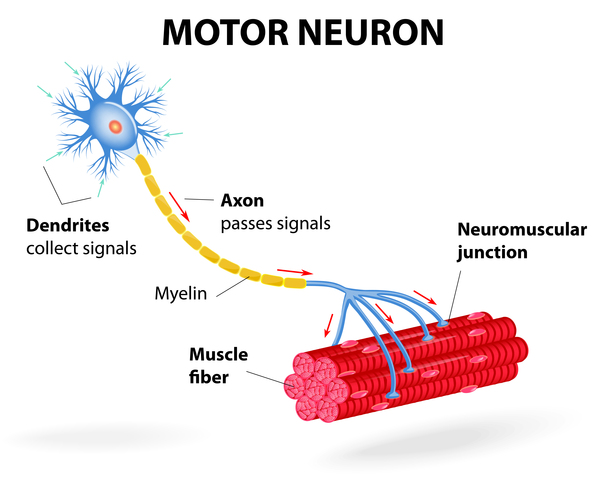
Some people with SCA36 have abnormalities of the eye muscles, which can lead to involuntary eye movements (nystagmus), rapid eye movements (saccades), trouble moving the eyes side-to-side (oculomotor apraxia), and droopy eyelids (ptosis). Sensorineural hearing loss, which is hearing loss caused by changes in the inner ear, may also occur in people with SCA36.

Brain imaging of people with SCA36 shows progressive atrophy of various parts of the brain, particularly within the cerebellum, which is the area of the brain involved in coordinating movements. Over time, the loss of cells in the cerebellum causes the movement problems characteristic of SCA36. In older affected individuals, the frontal lobes of the brain may show atrophy resulting in loss of executive function, which is the ability to plan and implement actions and develop problem-solving strategies.

Signs and symptoms of SCA36 typically begin in a person's forties or fifties but can appear anytime during adulthood. People with SCA36 have a normal lifespan and are usually mobile for 15 to 20 years after they are diagnosed.
Frequency
Approximately 100 individuals with SCA36 have been reported in the scientific literature. Almost all of these individuals have been from two regions: western Japan and the Costa de Morte in Galicia, Spain.
Causes
SCA36 is caused by mutations in the NOP56 gene. The NOP56 gene provides instructions for making a protein called nucleolar protein 56, which is primarily found in the nucleus of nerve cells (neurons), particularly those in the cerebellum. This protein is one part (subunit) of the ribonucleoprotein complex, which is composed of proteins and molecules of RNA, DNA's chemical cousin. The ribonucleoprotein complex is needed to make cellular structures called ribosomes, which process the cell's genetic instructions to create proteins.

The NOP56 gene mutations that cause SCA36 involve a string of six DNA building blocks (nucleotides) located in an area of the gene known as intron 1. This string of six nucleotides (known as a hexanucleotide) is represented by the letters GGCCTG and normally appears multiple times in a row. In healthy individuals, GGCCTG is repeated 3 to 14 times within the gene. In people with SCA36, GGCCTG is repeated at least 650 times. It is unclear if 15 to 649 repeats of this hexanucleotide cause any signs or symptoms.
To make proteins from the genetic instructions carried in genes, a molecule called messenger RNA (mRNA) is formed. This molecule acts as a genetic blueprint for protein production. However, a large increase in the number of GGCCTG repeats in the NOP56 gene disrupts the normal structure of NOP56 mRNA. Abnormal NOP56 mRNA molecules form clumps called RNA foci within the nucleus of neurons. Other proteins become trapped in the RNA foci, where they cannot function. These proteins may be important for controlling gene activity or protein production.
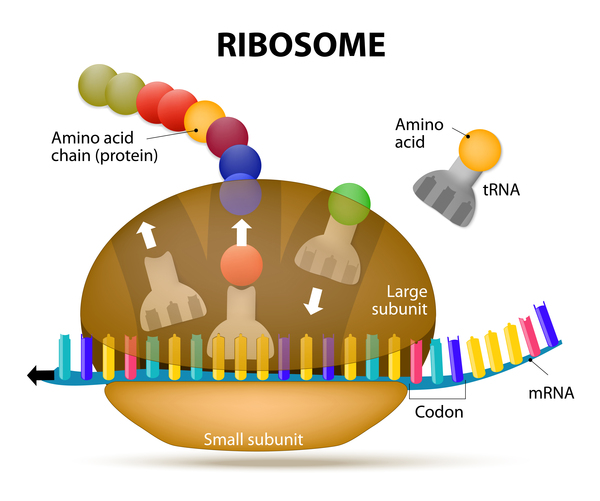
Additionally, researchers believe that the large expansion of the hexanucleotide repeat in the NOP56 gene may reduce the activity of a nearby gene called MIR1292. The MIR1292 gene provides instructions for making a type of RNA that regulates the activity (expression) of genes that produce proteins called glutamate receptors. These proteins are found on the surface of neurons and allow these cells to communicate with one another. A decrease in the production of Mir1292 RNA can lead to an increase in the production of glutamate receptors. The increased receptor activity may overexcite neurons, which disrupts normal communication between cells and can contribute to ataxia.
The combination of RNA foci and overly excited neurons likely leads to the death of these cells over time. Because the NOP56 gene is especially active in neurons in the cerebellum, these cells are particularly affected by expansion of the gene, leading to cerebellar atrophy. Deterioration in this part of the brain leads to ataxia and the other signs and symptoms of SCA36.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

In most cases, an affected person has one parent with the condition.
In conditions that are caused by repeated segments of DNA, the number of repeats often increases when the altered gene is passed down from one generation to the next. Additionally, a larger number of repeats is usually associated with an earlier onset of signs and symptoms. This phenomenon is called anticipation. Some families affected by SCA36 have demonstrated anticipation while others have not. When anticipation is observed in SCA36, the mutation is most often passed down from the affected father.
Other Names for This Condition
- Asidan ataxia
- Costa de Morte ataxia
- SCA36
- Spinocerebellar ataxia 36
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Abe K, Ikeda Y, Kurata T, Ohta Y, Manabe Y, Okamoto M, Takamatsu K, Ohta T, Takao Y, Shiro Y, Shoji M, Kamiya T, Kobayashi H, Koizumi A. Cognitive and affective impairments of a novel SCA/MND crossroad mutation Asidan. Eur J Neurol. 2012 Aug;19(8):1070-8. doi: 10.1111/j.1468-1331.2012.03669.x. Epub 2012 Feb 21. Citation on PubMed
- Garcia-Murias M, Quintans B, Arias M, Seixas AI, Cacheiro P, Tarrio R, Pardo J, Millan MJ, Arias-Rivas S, Blanco-Arias P, Dapena D, Moreira R, Rodriguez-Trelles F, Sequeiros J, Carracedo A, Silveira I, Sobrido MJ. 'Costa da Morte' ataxia is spinocerebellar ataxia 36: clinical and genetic characterization. Brain. 2012 May;135(Pt 5):1423-35. doi: 10.1093/brain/aws069. Epub 2012 Apr 3. Citation on PubMed or Free article on PubMed Central
- Ikeda Y, Ohta Y, Kobayashi H, Okamoto M, Takamatsu K, Ota T, Manabe Y, Okamoto K, Koizumi A, Abe K. Clinical features of SCA36: a novel spinocerebellar ataxia with motor neuron involvement (Asidan). Neurology. 2012 Jul 24;79(4):333-41. doi: 10.1212/WNL.0b013e318260436f. Epub 2012 Jun 27. Citation on PubMed
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011 Jul 15;89(1):121-30. doi: 10.1016/j.ajhg.2011.05.015. Epub 2011 Jun 16. Citation on PubMed or Free article on PubMed Central
- Liu W, Ikeda Y, Hishikawa N, Yamashita T, Deguchi K, Abe K. Characteristic RNA foci of the abnormal hexanucleotide GGCCUG repeat expansion in spinocerebellar ataxia type 36 (Asidan). Eur J Neurol. 2014 Nov;21(11):1377-86. doi: 10.1111/ene.12491. Epub 2014 Jul 2. Citation on PubMed
- Sugihara K, Maruyama H, Morino H, Miyamoto R, Ueno H, Matsumoto M, Kaji R, Kitaguchi H, Yukitake M, Higashi Y, Nishinaka K, Oda M, Izumi Y, Kawakami H. The clinical characteristics of spinocerebellar ataxia 36: a study of 2121 Japanese ataxia patients. Mov Disord. 2012 Aug;27(9):1158-63. doi: 10.1002/mds.25092. Epub 2012 Jul 2. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.