

Description
Spinocerebellar ataxia type 3 (SCA3) is a condition characterized by progressive problems with movement. People with this condition initially experience problems with coordination and balance (ataxia). Other early signs and symptoms of SCA3 include speech difficulties, uncontrolled muscle tensing (dystonia), muscle stiffness (spasticity), rigidity, tremors, bulging eyes, and double vision. People with this condition may experience sleep disorders such as restless leg syndrome or REM sleep behavior disorder. Restless leg syndrome is a condition characterized by numbness or tingling in the legs accompanied by an urge to move the legs to stop the sensations. REM sleep behavior disorder is a condition in which the muscles are active during the dream (REM) stage of sleep, so an affected person often acts out his or her dreams. These sleep disorders tend to leave affected individuals feeling tired during the day.
Over time, individuals with SCA3 may develop loss of sensation and weakness in the limbs (peripheral neuropathy), muscle cramps, muscle twitches (fasciculations), and swallowing difficulties. Individuals with SCA3 may have problems with memory, planning, and problem solving.
Signs and symptoms of the disorder typically begin in mid-adulthood but can appear anytime from childhood to late adulthood. People with SCA3 eventually require wheelchair assistance. They usually survive 10 to 20 years after symptoms first appear.
Frequency
The prevalence of SCA3 is unknown. This condition is thought to be the most common type of spinocerebellar ataxia; however, all types of spinocerebellar ataxia are relatively rare. SCA3 appears to be more common in certain populations, such as some Indigenous (native) Australian populations.
Causes
Mutations in the ATXN3 gene cause SCA3. The ATXN3 gene provides instructions for making an enzyme called ataxin-3, which is found in cells throughout the body. Ataxin-3 is involved in a mechanism called the ubiquitin-proteasome system that destroys and gets rid of excess or damaged proteins. The molecule ubiquitin is attached (bound) to unneeded proteins, which tags them to be broken down (degraded) within cells. Ataxin-3 removes the ubiquitin from these unwanted proteins just before they are degraded so that the ubiquitin can be used again. Researchers believe that ataxin-3 also may be involved in regulating the first stage of protein production (transcription).
The ATXN3 gene mutations that cause SCA3 involve a DNA segment known as a CAG trinucleotide repeat. This segment is made up of a series of three DNA building blocks (cytosine, adenine, and guanine) that appear multiple times in a row. Normally, the CAG segment is repeated 12 to 43 times within the gene. Most people have fewer than 31 CAG repeats. In people with SCA3, the CAG segment is repeated more than 50 times. People who have 44 to 52 CAG repeats are described as having an "intermediate repeat." These individuals may or may not develop SCA3. People with 75 or fewer repeats tend to first experience signs and symptoms of SCA3 in mid-adulthood, while people with around 80 repeats usually have signs and symptoms by their teens.
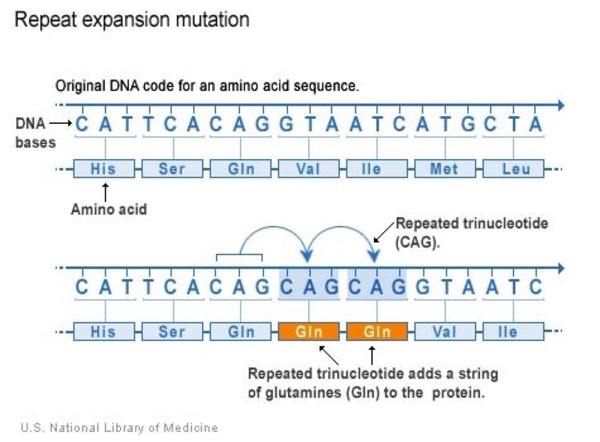
An increase in the length of the CAG segment leads to the production of an abnormally long version of the ataxin-3 enzyme that folds into the wrong 3-dimensional shape. This nonfunctional ataxin-3 enzyme cannot remove ubiquitin from proteins that are no longer needed. As a result, these unwanted proteins, along with ubiquitin and ataxin-3, cluster together to form clumps (aggregates) within the nucleus of the cells. It is unclear how these aggregates affect cell function, because they are found in healthy cells as well as those that die.
Nerve cells (neurons) and other types of brain cells are most affected by mutations in the ATXN3 gene. SCA3 is associated with cell death in the part of the brain that is connected to the spinal cord (the brainstem), the part of the brain involved in coordinating movements (the cerebellum), and other areas of the brain. This condition is also associated with the death of neurons in the spinal cord. Over time, the loss of cells in the brain and spinal cord cause the signs and symptoms characteristic of SCA3.

Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.

As the altered ATXN3 gene is passed down from one generation to the next, the length of the CAG trinucleotide repeat often increases. A larger number of repeats is usually associated with an earlier onset and faster progression of signs and symptoms. This phenomenon is called anticipation. Anticipation may be more prominent when the ATXN3 gene is inherited from a person's father (paternal inheritance) than when it is inherited from a person's mother (maternal inheritance).
In rare cases, individuals have been reported with expanded CAG repeats on both copies of the ATXN3 gene in each cell. These people tend to have more severe signs and symptoms than people with only one mutation, and features of the condition appear in childhood.
Other Names for This Condition
- Azorean ataxia
- Azorean disease
- Machado-Joseph disease
- MJD
- SCA3
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Camey S, Jardim LB, Kieling C, Saute JA, Vigo A. A prospective study of SCA3 gait ataxia described through a Markovian method. Neuroepidemiology. 2010;34(3):163-70. doi: 10.1159/000279333. Epub 2010 Feb 2. Citation on PubMed
- D'Abreu A, Franca MC Jr, Paulson HL, Lopes-Cendes I. Caring for Machado-Joseph disease: current understanding and how to help patients. Parkinsonism Relat Disord. 2010 Jan;16(1):2-7. doi: 10.1016/j.parkreldis.2009.08.012. Epub 2009 Oct 6. Citation on PubMed or Free article on PubMed Central
- Kieling C, Prestes PR, Saraiva-Pereira ML, Jardim LB. Survival estimates for patients with Machado-Joseph disease (SCA3). Clin Genet. 2007 Dec;72(6):543-5. doi: 10.1111/j.1399-0004.2007.00910.x. Epub 2007 Sep 25. Citation on PubMed
- Liang X, Jiang H, Chen C, Zhou G, Wang J, Zhang S, Lei L, Wang X, Tang B. The correlation between magnetic resonance imaging features of the brainstem and cerebellum and clinical features of spinocerebellar ataxia 3/Machado-Joseph disease. Neurol India. 2009 Sep-Oct;57(5):578-83. doi: 10.4103/0028-3886.57803. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.