

Description
Spinocerebellar ataxia type 1 (SCA1) is a condition characterized by progressive problems with movement. People with this condition initially experience problems with coordination and balance (ataxia). Other signs and symptoms of SCA1 include speech and swallowing difficulties, muscle stiffness (spasticity), and weakness in the muscles that control eye movement (ophthalmoplegia). Eye muscle weakness leads to rapid, involuntary eye movements (nystagmus). Individuals with SCA1 may have difficulty processing, learning, and remembering information (cognitive impairment).
Over time, individuals with SCA1 may develop numbness, tingling, or pain in the arms and legs (sensory neuropathy); uncontrolled muscle tensing (dystonia); muscle wasting (atrophy); and muscle twitches (fasciculations). Rarely, rigidity, tremors, and involuntary jerking movements (chorea) have been reported in people who have been affected for many years.
Signs and symptoms of the disorder typically begin in early adulthood but can appear anytime from childhood to late adulthood. People with SCA1 typically survive 10 to 20 years after symptoms first appear.
Frequency
SCA1 affects 1 to 2 per 100,000 people worldwide.
Causes
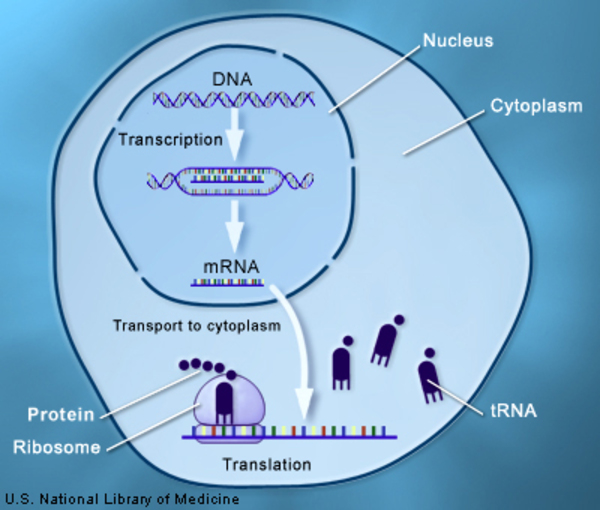
Mutations in the ATXN1 gene cause SCA1. The ATXN1 gene provides instructions for making a protein called ataxin-1. This protein is found throughout the body, but its function is unknown. Within cells, ataxin-1 is located in the nucleus. Researchers believe that ataxin-1 may be involved in regulating various aspects of producing proteins, including the first stage of protein production (transcription) and processing RNA, a chemical cousin of DNA.
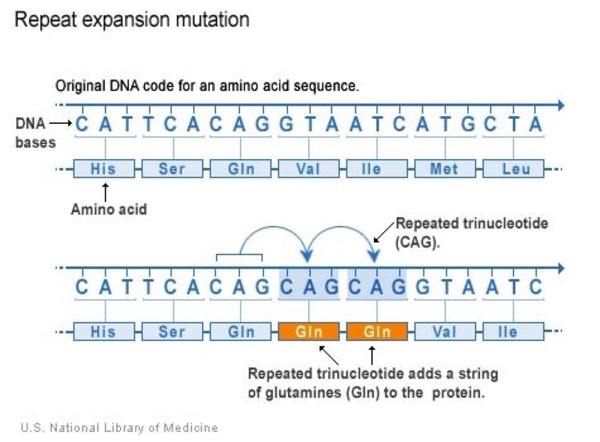
The ATXN1 gene mutations that cause SCA1 involve a DNA segment known as a CAG trinucleotide repeat. This segment is made up of a series of three DNA building blocks (cytosine, adenine, and guanine) that appear multiple times in a row. Normally, the CAG segment is repeated 4 to 39 times within the gene. In people with SCA1, the CAG segment is repeated 40 to more than 80 times. People with 40 to 50 repeats tend to first experience signs and symptoms of SCA1 in mid-adulthood, while people with more than 70 repeats usually have signs and symptoms by their teens.
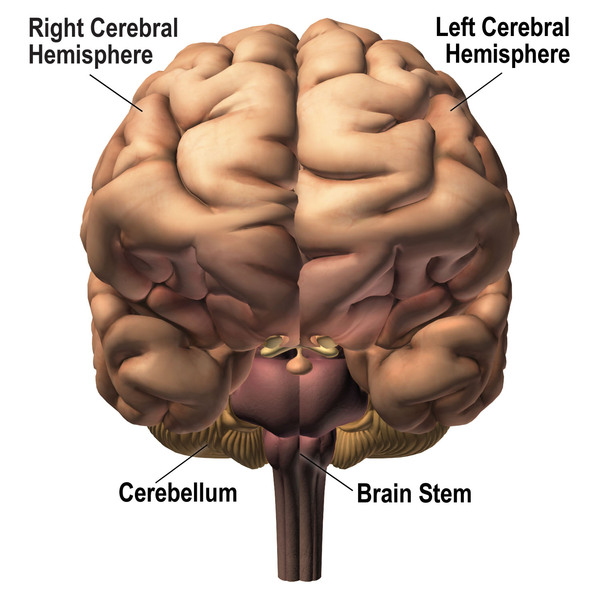
An increase in the length of the CAG segment leads to the production of an abnormally long version of the ataxin-1 protein that folds into the wrong 3-dimensional shape. This abnormal protein clusters with other proteins to form clumps (aggregates) within the nucleus of the cells. These aggregates prevent the ataxin-1 protein from functioning normally, which damages cells and leads to cell death. For reasons that are unclear, aggregates of ataxin-1 are found only in the brain and spinal cord (central nervous system). Cells within the cerebellum, which is the part of the brain that coordinates movement, are particularly sensitive to changes in ataxin-1 shape and function. Over time, the loss of the cells of the cerebellum causes the movement problems characteristic of SCA1.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. An affected person usually inherits the altered gene from one affected parent. However, some people with SCA1 do not have a parent with the disorder.

As the altered ATXN1 gene is passed down from one generation to the next, the length of the CAG trinucleotide repeat often increases. A larger number of repeats is usually associated with an earlier onset of signs and symptoms. This phenomenon is called anticipation. Anticipation tends to be more prominent when the ATXN1 gene is inherited from a person's father (paternal inheritance) than when it is inherited from a person's mother (maternal inheritance).
Individuals who have around 35 CAG repeats in the ATXN1 gene do not develop SCA1, but they are at risk of having children who will develop the disorder. As the gene is passed from parent to child, the size of the CAG trinucleotide repeat may lengthen into the range associated with SCA1 (40 repeats or more).
Other Names for This Condition
- Olivopontocerebellar atrophy I
- SCA1
- Spinocerebellar atrophy I
- Type 1 spinocerebellar ataxia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Globas C, du Montcel ST, Baliko L, Boesch S, Depondt C, DiDonato S, Durr A, Filla A, Klockgether T, Mariotti C, Melegh B, Rakowicz M, Ribai P, Rola R, Schmitz-Hubsch T, Szymanski S, Timmann D, Van de Warrenburg BP, Bauer P, Schols L. Early symptoms in spinocerebellar ataxia type 1, 2, 3, and 6. Mov Disord. 2008 Nov 15;23(15):2232-8. doi: 10.1002/mds.22288. Citation on PubMed
- Kang S, Hong S. Molecular pathogenesis of spinocerebellar ataxia type 1 disease. Mol Cells. 2009 Jun 30;27(6):621-7. doi: 10.1007/s10059-009-0095-y. Epub 2009 Jun 22. Citation on PubMed
- Matilla-Duenas A, Goold R, Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum. 2008;7(2):106-14. doi: 10.1007/s12311-008-0009-0. Citation on PubMed
- Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem. 2009 Mar 20;284(12):7425-9. doi: 10.1074/jbc.R800041200. Epub 2008 Oct 28. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.