

Description
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is an inherited condition that causes muscle weakness and respiratory failure typically beginning in infancy. Early features of this condition are difficult and noisy breathing, especially when inhaling; a weak cry; problems feeding; and recurrent episodes of pneumonia. Typically between the ages of 6 weeks and 6 months, infants with this condition will experience a sudden inability to breathe due to paralysis of the muscle that separates the abdomen from the chest cavity (the diaphragm). Normally, the diaphragm contracts and moves downward during inhalation to allow the lungs to expand. With diaphragm paralysis, affected individuals require life-long support with a machine to help them breathe (mechanical ventilation). Rarely, children with SMARD1 develop signs or symptoms of the disorder later in childhood.
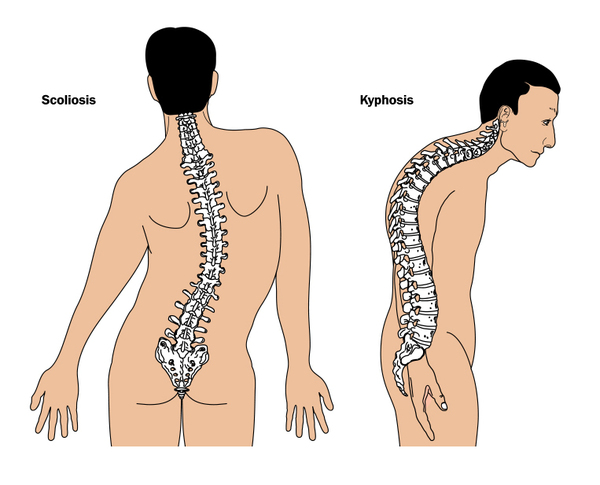
Soon after respiratory failure occurs, individuals with SMARD1 develop muscle weakness in their distal muscles. These are the muscles farther from the center of the body, such as muscles in the hands and feet. The weakness soon spreads to all muscles; however, within 2 years, the muscle weakness typically stops getting worse. Some individuals may retain a low level of muscle function, while others lose all ability to move their muscles. Muscle weakness severely impairs motor development, such as sitting, standing, and walking. Some affected children develop an abnormal side-to-side and back-to-front curvature of the spine (scoliosis and kyphosis, often called kyphoscoliosis when they occur together). After approximately the first year of life, individuals with SMARD1 may lose their deep tendon reflexes, such as the reflex being tested when a doctor taps the knee with a hammer.
Other features of SMARD1 can include reduced pain sensitivity, excessive sweating (hyperhidrosis), loss of bladder and bowel control, and an irregular heartbeat (arrhythmia).
Frequency
SMARD1 appears to be a rare condition, but its prevalence is unknown. More than 60 cases have been reported in the scientific literature.
Causes
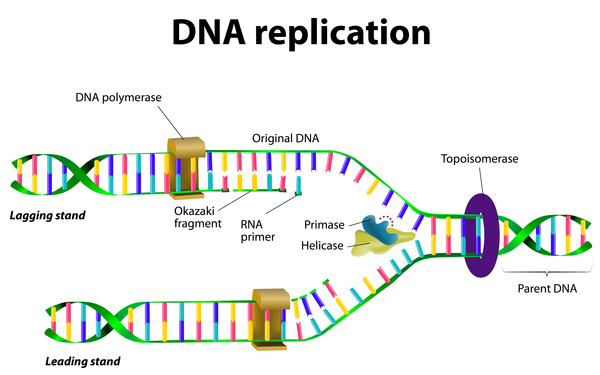
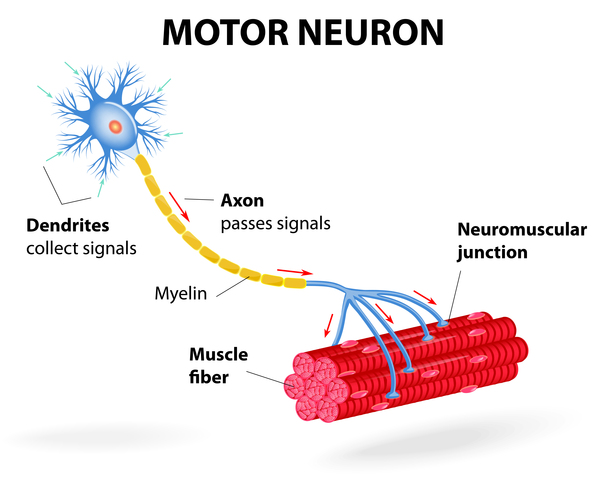
Mutations in the IGHMBP2 gene cause SMARD1. The IGHMBP2 gene provides instructions for making a protein involved in copying (replicating) DNA; producing RNA, a chemical cousin of DNA; and producing proteins. IGHMBP2 gene mutations that cause SMARD1 lead to the production of a protein with reduced ability to aid in DNA replication and the production of RNA and proteins. These problems particularly affect alpha-motor neurons, which are specialized cells in the brainstem and spinal cord that control muscle movements. Although the mechanism is unknown, altered IGHMBP2 proteins contribute to the damage of these neurons and their death over time. The cumulative death of alpha-motor neurons leads to breathing problems and progressive muscle weakness in children with SMARD1.
Research suggests that the amount of functional protein that is produced from the mutated IGHMBP2 gene may play a role in the severity of SMARD1. Individuals who have some functional protein are more likely to develop signs and symptoms later in childhood and retain a greater level of muscle function.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Autosomal recessive distal spinal muscular atrophy 1
- DHMN6
- Diaphragmatic spinal muscular atrophy
- Distal hereditary motor neuronopathy type VI
- Distal spinal muscular atrophy type 1
- DSMA1
- HMN6
- HMNVI
- Severe infantile axonal neuropathy with respiratory failure
- SIANRF
- SMARD1
- Spinal muscular atrophy with respiratory distress
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Eckart M, Guenther UP, Idkowiak J, Varon R, Grolle B, Boffi P, Van Maldergem L, Hubner C, Schuelke M, von Au K. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics. 2012 Jan;129(1):e148-56. doi: 10.1542/peds.2011-0544. Epub 2011 Dec 12. Citation on PubMed
- Guenther UP, Handoko L, Varon R, Stephani U, Tsao CY, Mendell JR, Lutzkendorf S, Hubner C, von Au K, Jablonka S, Dittmar G, Heinemann U, Schuetz A, Schuelke M. Clinical variability in distal spinal muscular atrophy type 1 (DSMA1): determination of steady-state IGHMBP2 protein levels in five patients with infantile and juvenile disease. J Mol Med (Berl). 2009 Jan;87(1):31-41. doi: 10.1007/s00109-008-0402-7. Epub 2008 Sep 18. Citation on PubMed
- Guenther UP, Varon R, Schlicke M, Dutrannoy V, Volk A, Hubner C, von Au K, Schuelke M. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis. Hum Mutat. 2007 Aug;28(8):808-15. doi: 10.1002/humu.20525. Citation on PubMed
- Kaindl AM, Guenther UP, Rudnik-Schoneborn S, Varon R, Zerres K, Schuelke M, Hubner C, von Au K. Spinal muscular atrophy with respiratory distress type 1 (SMARD1). J Child Neurol. 2008 Feb;23(2):199-204. doi: 10.1177/0883073807310989. Citation on PubMed
- van der Pol WL, Talim B, Pitt M, von Au K. 190 th ENMC international workshop: Spinal muscular atrophy with respiratory distress/distal spinal muscular atrophy type 1: 11-13 May 2012, Naarden, The Netherlands. Neuromuscul Disord. 2013 Jul;23(7):602-9. doi: 10.1016/j.nmd.2013.04.004. Epub 2013 May 29. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.