

Description
Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME) is a neurological condition that begins in childhood. SMA-PME causes muscle weakness and wasting (atrophy) and a combination of seizures and uncontrollable muscle jerks (myoclonic epilepsy).
In individuals with SMA-PME, spinal muscular atrophy results from a loss of specialized nerve cells, called motor neurons, in the spinal cord and the part of the brain that is connected to the spinal cord (the brainstem ). After a few years of normal development, affected children begin experiencing muscle weakness and atrophy in the lower limbs, causing difficulty walking and frequent falls. The muscles in the upper limbs are later affected, and soon the muscle weakness and atrophy spreads throughout the body. Once weakness reaches the muscles used for breathing and swallowing, affected individuals develop life-threatening breathing problems and increased susceptibility to pneumonia.
). After a few years of normal development, affected children begin experiencing muscle weakness and atrophy in the lower limbs, causing difficulty walking and frequent falls. The muscles in the upper limbs are later affected, and soon the muscle weakness and atrophy spreads throughout the body. Once weakness reaches the muscles used for breathing and swallowing, affected individuals develop life-threatening breathing problems and increased susceptibility to pneumonia.
A few years after the muscle weakness begins, affected individuals start to experience recurrent seizures (epilepsy). Most people with SMA-PME have a variety of seizure types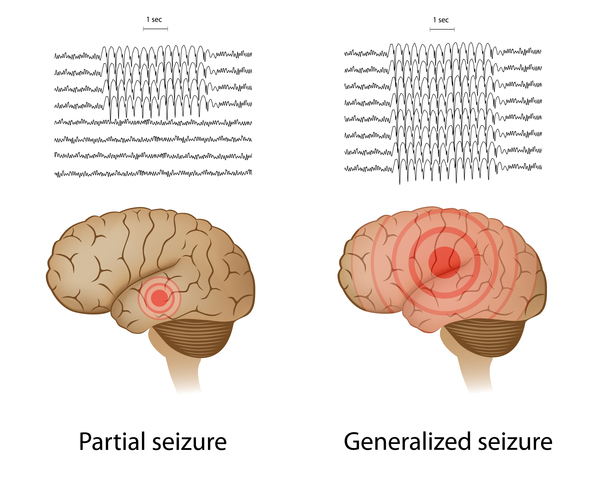 . In addition to myoclonic epilepsy, they may have generalized tonic-clonic seizures (also known as grand mal seizures), which cause muscle rigidity, convulsions, and loss of consciousness. Affected individuals can also have absence seizures, which cause loss of consciousness for a short period that may or may not be accompanied by muscle jerks. In SMA-PME, seizures often increase in frequency over time and are usually not well-controlled with medication. Individuals with SMA-PME may also have episodes of rhythmic shaking (tremors), usually in the hands; these tremors are not thought to be related to epilepsy. Some people with SMA-PME develop hearing loss caused by nerve damage in the inner ear
. In addition to myoclonic epilepsy, they may have generalized tonic-clonic seizures (also known as grand mal seizures), which cause muscle rigidity, convulsions, and loss of consciousness. Affected individuals can also have absence seizures, which cause loss of consciousness for a short period that may or may not be accompanied by muscle jerks. In SMA-PME, seizures often increase in frequency over time and are usually not well-controlled with medication. Individuals with SMA-PME may also have episodes of rhythmic shaking (tremors), usually in the hands; these tremors are not thought to be related to epilepsy. Some people with SMA-PME develop hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss).
(sensorineural hearing loss).
Individuals with SMA-PME have a shortened lifespan; they generally live into late childhood or early adulthood. Near the end of their lives, affected individuals often have limited mobility, difficulty swallowing, and decline in cognitive functioning. The cause of death is often respiratory failure or pneumonia.
Frequency
SMA-PME is a rare disorder; approximately 50 affected individuals have been described in the scientific literature.
Causes
SMA-PME is caused by variants (also called mutations) in the ASAH1 gene. This gene provides instructions for making an enzyme called acid ceramidase. This enzyme is found in lysosomes , which are cell compartments that digest and recycle materials. Within lysosomes, acid ceramidase breaks down fats called ceramides into a fat called sphingosine and a fatty acid. These two breakdown products are recycled to create new ceramides for the body to use. Ceramides have several roles within cells. For example, they are a component of a fatty substance called myelin
, which are cell compartments that digest and recycle materials. Within lysosomes, acid ceramidase breaks down fats called ceramides into a fat called sphingosine and a fatty acid. These two breakdown products are recycled to create new ceramides for the body to use. Ceramides have several roles within cells. For example, they are a component of a fatty substance called myelin that insulates and protects nerve cells.
that insulates and protects nerve cells.
ASAH1 gene variants that cause SMA-PME result in a reduction of acid ceramidase activity to a level less than one-third of normal. Inefficient breakdown of ceramides and impaired production of its breakdown products likely play a role in the nerve cell damage that leads to the features of SMA-PME, but the exact mechanism is unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Hereditary myoclonus with progressive distal muscular atrophy
- Jankovic-Rivera syndrome
- SMA-PME
- SMAPME
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dyment DA, Sell E, Vanstone MR, Smith AC, Garandeau D, Garcia V, Carpentier S, Le Trionnaire E, Sabourdy F, Beaulieu CL, Schwartzentruber JA, McMillan HJ; FORGE Canada Consortium; Majewski J, Bulman DE, Levade T, Boycott KM. Evidence for clinical, genetic and biochemical variability in spinal muscular atrophy with progressive myoclonic epilepsy. Clin Genet. 2014 Dec;86(6):558-63. doi: 10.1111/cge.12307. Epub 2013 Nov 21. Citation on PubMed
- Striano P, Boccella P, Sarappa C, Striano S. Spinal muscular atrophy and progressive myoclonic epilepsy: one case report and characteristics of the epileptic syndrome. Seizure. 2004 Dec;13(8):582-6. doi: 10.1016/j.seizure.2004.01.008. Citation on PubMed
- Yu FPS, Amintas S, Levade T, Medin JA. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis. 2018 Jul 20;13(1):121. doi: 10.1186/s13023-018-0845-z. Citation on PubMed
- Zhou J, Tawk M, Tiziano FD, Veillet J, Bayes M, Nolent F, Garcia V, Servidei S, Bertini E, Castro-Giner F, Renda Y, Carpentier S, Andrieu-Abadie N, Gut I, Levade T, Topaloglu H, Melki J. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am J Hum Genet. 2012 Jul 13;91(1):5-14. doi: 10.1016/j.ajhg.2012.05.001. Epub 2012 Jun 14. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.