

Description
Spinal muscular atrophy is a genetic disorder characterized by weakness and wasting (atrophy) in muscles used for movement (skeletal muscles). It is caused by a loss of specialized nerve cells, called motor neurons that control muscle movement. The weakness tends to be more severe in the muscles that are close to the center of the body (proximal) compared to muscles away from the body's center (distal). The muscle weakness usually worsens with age. There are many types of spinal muscular atrophy that are caused by changes in the same genes. The types differ in age of onset and severity of muscle weakness; however, there is overlap between the types. Other forms of spinal muscular atrophy and related motor neuron diseases, such as spinal muscular atrophy with progressive myoclonic epilepsy, spinal muscular atrophy with lower extremity predominance, X-linked infantile spinal muscular atrophy, and spinal muscular atrophy with respiratory distress type 1 are caused by mutations in other genes.
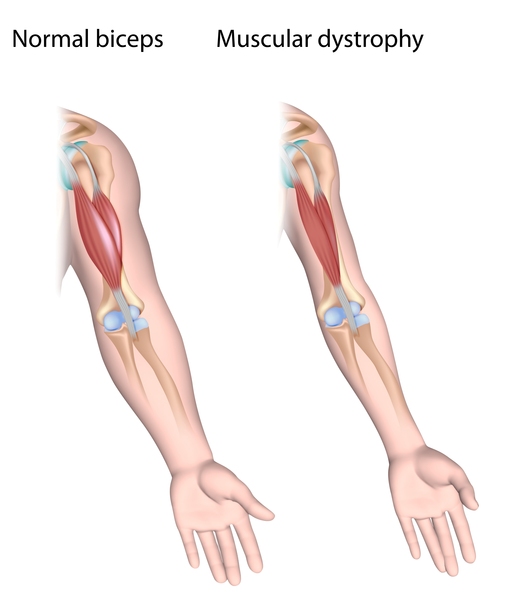
Spinal muscular atrophy type 0 is evident before birth and is the rarest and most severe form of the condition. Affected infants move less in the womb, and as a result they are often born with joint deformities (contractures). They have extremely weak muscle tone (hypotonia) at birth. Their respiratory muscles are very weak and they often do not survive past infancy due to respiratory failure. Some infants with spinal muscular atrophy type 0 also have heart defects that are present from birth (congenital).
Spinal muscular atrophy type I (also called Werdnig-Hoffmann disease) is the most common form of the condition. It is a severe form of the disorder with muscle weakness evident at birth or within the first few months of life. Most affected children cannot control their head movements or sit unassisted. Children with this type may have swallowing problems that can lead to difficulty feeding and poor growth. They can also have breathing problems due to weakness of respiratory muscles and an abnormally bell-shaped chest that prevents the lungs from fully expanding. Most children with spinal muscular atrophy type I do not survive past early childhood due to respiratory failure.
Spinal muscular atrophy type II (also called Dubowitz disease) is characterized by muscle weakness that develops in children between ages 6 and 12 months. Children with this type can sit without support, although they may need help getting to a seated position. However, as the muscle weakness worsens later in childhood, affected individuals may need support to sit. Individuals with spinal muscular atrophy type II cannot stand or walk unaided. They often have involuntary trembling (tremors) in their fingers, a spine that curves side-to-side (scoliosis), and respiratory muscle weakness that can be life-threatening. The life span of individuals with spinal muscular atrophy type II varies, but many people with this condition live into their twenties or thirties.

Spinal muscular atrophy type III (also called Kugelberg-Welander disease) typically causes muscle weakness after early childhood. Individuals with this condition can stand and walk unaided, but over time, walking and climbing stairs may become increasingly difficult. Many affected individuals require wheelchair assistance later in life. People with spinal muscular atrophy type III typically have a normal life expectancy.
Spinal muscular atrophy type IV is rare and often begins in early adulthood. Affected individuals usually experience mild to moderate muscle weakness, tremors, and mild breathing problems. People with spinal muscular atrophy type IV have a normal life expectancy.
Frequency
Spinal muscular atrophy affects 1 per 8,000 to 10,000 people worldwide. Spinal muscular atrophy type I is the most common type, accounting for about half of all cases. Types II and III are the next most common and types 0 and IV are rare.
Causes
Mutations in the SMN1 gene cause all types of spinal muscular atrophy described above. The number of copies of the SMN2 gene modifies the severity of the condition and helps determine which type develops.
The SMN1 and SMN2 genes both provide instructions for making a protein called the survival motor neuron (SMN) protein. Normally, most functional SMN protein is produced from the SMN1 gene, with a small amount produced from the SMN2 gene. Several different versions of the SMN protein are produced from the SMN2 gene, but only one version is functional; the other versions are smaller and quickly broken down. The SMN protein is one of a group of proteins called the SMN complex, which is important for the maintenance of motor neurons. Motor neurons transmit signals from the brain and spinal cord that tell skeletal muscles to tense (contract), which allows the body to move.
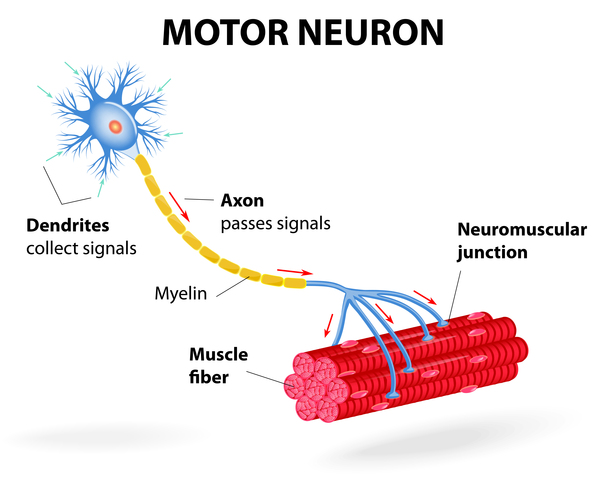
Most people with spinal muscular atrophy are missing a piece of the SMN1 gene, which impairs SMN protein production. A shortage of SMN protein leads to motor neuron death, and as a result, signals are not transmitted between the brain and muscles. Muscles cannot contract without receiving signals from the brain, so many skeletal muscles become weak and waste away, leading to the signs and symptoms of spinal muscular atrophy.
Typically, people have two copies of the SMN1 gene and one to two copies of the SMN2 gene in each cell. However, the number of copies of the SMN2 gene varies, with some people having up to eight copies. In people with spinal muscular atrophy, having multiple copies of the SMN2 gene is usually associated with less severe features of the condition that develop later in life. The SMN protein produced by the SMN2 genes can help make up for the protein deficiency caused by SMN1 gene mutations. People with spinal muscular atrophy type 0 usually have one copy of the SMN2 gene in each cell, while those with type I generally have one or two copies, those with type II usually have three copies, those with type III have three or four copies, and those with type IV have four or more copies. Other factors, many unknown, also contribute to the variable severity of spinal muscular atrophy.
Inheritance
Spinal muscular atrophy is inherited in an autosomal recessive pattern, which means both copies of the SMN1 gene in each cell have mutations. In most cases, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. In rare cases, a person with spinal muscular atrophy inherits an SMN1 gene mutation from one parent and acquires a new mutation in the other copy of the gene that occurs during the formation of reproductive cells (eggs or sperm) or in early embryonic development. In these cases, only one parent is a carrier of the SMN1 gene mutation.

Individuals who have more than the usual two copies of the SMN2 gene usually do not inherit the extra copies from a parent. They typically arise during a random error when making new copies of DNA (replication) in an egg or sperm cell or just after fertilization.
Other Names for This Condition
- 5q SMA
- Proximal SMA
- SMA
- SMA-associated SMA
- Spinal amyotrophies
- Spinal amyotrophy
- Spinal muscle degeneration
- Spinal muscle wasting
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Farrar MA, Kiernan MC. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics. 2015 Apr;12(2):290-302. doi: 10.1007/s13311-014-0314-x. Citation on PubMed or Free article on PubMed Central
- Fuller HR, Gillingwater TH, Wishart TM. Commonality amid diversity: Multi-study proteomic identification of conserved disease mechanisms in spinal muscular atrophy. Neuromuscul Disord. 2016 Sep;26(9):560-9. doi: 10.1016/j.nmd.2016.06.004. Epub 2016 Jun 7. Citation on PubMed
- Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin. 2015 Nov;33(4):831-46. doi: 10.1016/j.ncl.2015.07.004. Citation on PubMed or Free article on PubMed Central
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008 Jun 21;371(9630):2120-33. doi: 10.1016/S0140-6736(08)60921-6. Citation on PubMed
- Prior TW, Leach ME, Finanger E. Spinal Muscular Atrophy. 2000 Feb 24 [updated 2020 Dec 3]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1352/ Citation on PubMed
- Prior TW, Nagan N. Spinal Muscular Atrophy: Overview of Molecular Diagnostic Approaches. Curr Protoc Hum Genet. 2016 Jan 1;88:9.27.1-9.27.13. doi: 10.1002/0471142905.hg0927s88. Citation on PubMed
- Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A. 2004 Oct 15;130A(3):307-10. doi: 10.1002/ajmg.a.30251. Citation on PubMed or Free article on PubMed Central
- Wirth B, Brichta L, Schrank B, Lochmuller H, Blick S, Baasner A, Heller R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. 2006 May;119(4):422-8. doi: 10.1007/s00439-006-0156-7. Epub 2006 Mar 1. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.