

Description
Spastic paraplegia type 7 (also called SPG7) is one of more than 80 genetic disorders known as hereditary spastic paraplegias. These disorders primarily affect the brain and spinal cord (central nervous system), specifically nerve cells (neurons) that extend down the spinal cord. These neurons are used for muscle movement and sensation. Signs and symptoms of hereditary spastic paraplegias are characterized by progressive muscle stiffness (spasticity) in the legs and difficulty walking.
Hereditary spastic paraplegias are divided into two types: pure and complex. The pure types generally involve only spasticity of the lower limbs and walking difficulties. The complex types involve more widespread problems with the nervous system; the structure or functioning of the brain; and the nerves connecting the brain and spinal cord to muscles and sensory cells that detect sensations such as touch, pain, heat, and sound (the peripheral nervous system). In complex forms, there can also be features outside of the nervous system. Spastic paraplegia type 7 can occur in either the pure or complex form.
Like all hereditary spastic paraplegias, spastic paraplegia type 7 involves spasticity of the leg muscles and some muscle weakness. People with this form of spastic paraplegia can also have ataxia; a pattern of movement abnormalities known as parkinsonism; exaggerated reflexes (hyperreflexia) in the arms; speech difficulties (dysarthria); difficulty swallowing (dysphagia); involuntary movements of the eyes (nystagmus); mild hearing loss; abnormal curvature of the spine (scoliosis ); high-arched feet (pes cavus
); high-arched feet (pes cavus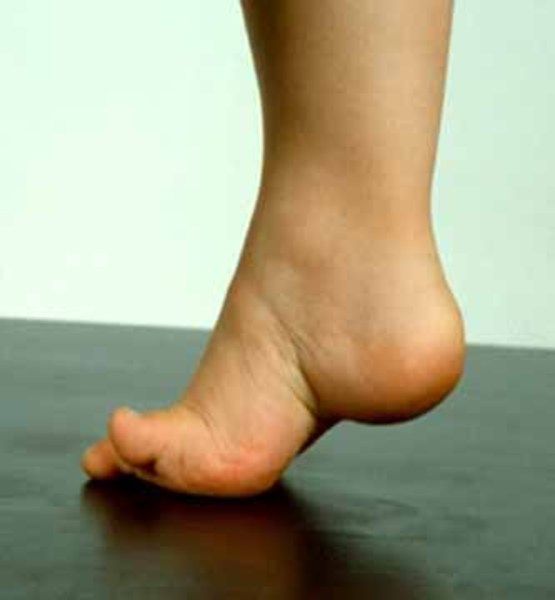 ); numbness, tingling, or pain in the arms and legs (sensory neuropathy); disturbance in the nerves used for muscle movement
); numbness, tingling, or pain in the arms and legs (sensory neuropathy); disturbance in the nerves used for muscle movement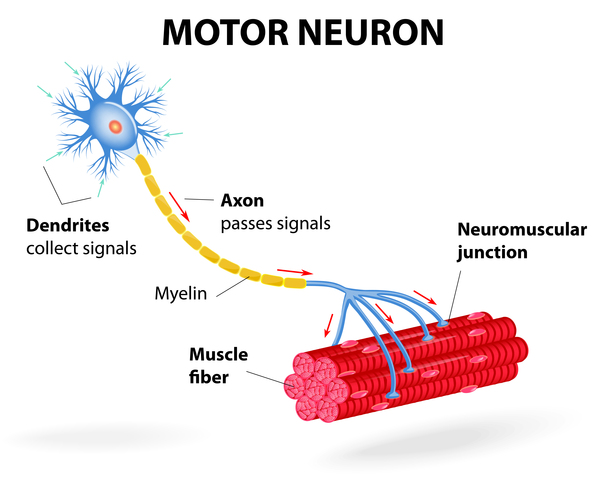 (motor neuropathy); and muscle wasting (amyotrophy). The onset of symptoms varies greatly among those with spastic paraplegia type 7; however, abnormalities in muscle tone and other features usually become noticeable in adulthood.
(motor neuropathy); and muscle wasting (amyotrophy). The onset of symptoms varies greatly among those with spastic paraplegia type 7; however, abnormalities in muscle tone and other features usually become noticeable in adulthood.
Frequency
The prevalence of all hereditary spastic paraplegias combined is estimated to be 2 to 6 in 100,000 people worldwide. This group of conditions is the most common cause of inherited spasticity. Spastic paraplegia type 7 likely accounts for only a small percentage of all spastic paraplegia cases.
Causes
Mutations in the SPG7 gene cause spastic paraplegia type 7. The SPG7 gene provides instructions for producing a protein called paraplegin. Located within the inner membrane of the energy-producing centers of cells (mitochondria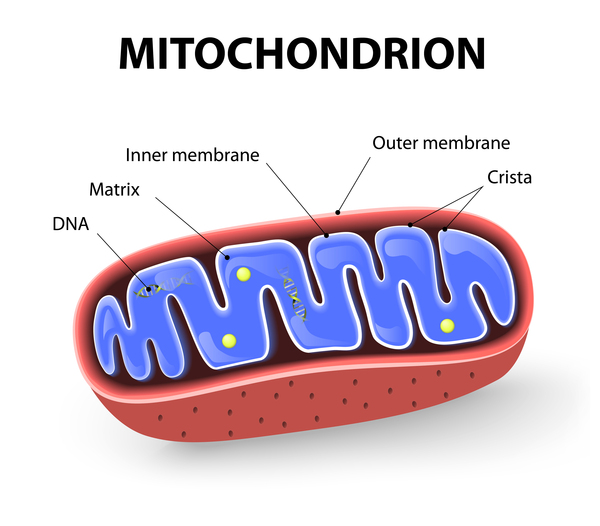 ), paraplegin is one of the proteins that form a complex called the m-AAA protease. The m-AAA protease acts as an enzyme and is responsible for assembling ribosomes
), paraplegin is one of the proteins that form a complex called the m-AAA protease. The m-AAA protease acts as an enzyme and is responsible for assembling ribosomes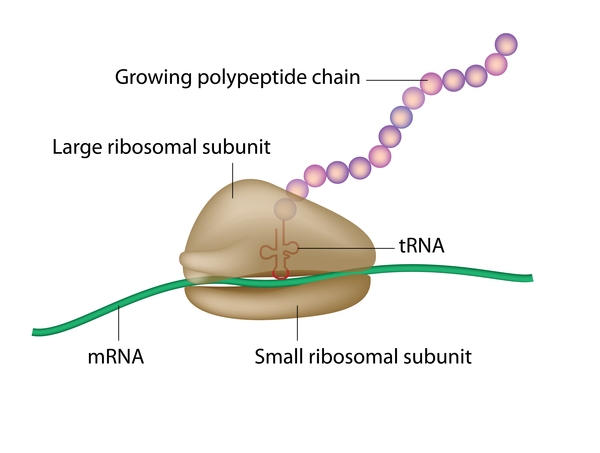 (cellular structures that process the cell's genetic instructions to create proteins) and removing nonfunctional proteins in the mitochondria.
(cellular structures that process the cell's genetic instructions to create proteins) and removing nonfunctional proteins in the mitochondria.
When there is a mutation in the SPG7 gene, the m-AAA protease cannot function correctly. Nonfunctional m-AAA proteases cause a build-up of unusable proteins in the mitochondria of nerve cells, which can result in swelling of the cell, reduced cell signaling, and impaired cell movement, leading to the major signs and symptoms of spastic paraplegia type 7.
Inheritance
In most cases, spastic paraplegia type 7 is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
In rare cases, spastic paraplegia type 7 is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Hereditary spastic paraplegia, paraplegin type
- Spastic paraplegia 7
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Atorino L, Silvestri L, Koppen M, Cassina L, Ballabio A, Marconi R, Langer T, Casari G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol. 2003 Nov 24;163(4):777-87. doi: 10.1083/jcb.200304112. Epub 2003 Nov 17. Citation on PubMed or Free article on PubMed Central
- Coarelli G, Schule R, van de Warrenburg BPC, De Jonghe P, Ewenczyk C, Martinuzzi A, Synofzik M, Hamer EG, Baets J, Anheim M, Schols L, Deconinck T, Masrori P, Fontaine B, Klockgether T, D'Angelo MG, Monin ML, De Bleecker J, Migeotte I, Charles P, Bassi MT, Klopstock T, Mochel F, Ollagnon-Roman E, D'Hooghe M, Kamm C, Kurzwelly D, Papin M, Davoine CS, Banneau G, Tezenas du Montcel S, Seilhean D, Brice A, Duyckaerts C, Stevanin G, Durr A. Loss of paraplegin drives spasticity rather than ataxia in a cohort of 241 patients with SPG7. Neurology. 2019 Jun 4;92(23):e2679-e2690. doi: 10.1212/WNL.0000000000007606. Epub 2019 May 8. Citation on PubMed or Free article on PubMed Central
- Hewamadduma CA, Hoggard N, O'Malley R, Robinson MK, Beauchamp NJ, Segamogaite R, Martindale J, Rodgers T, Rao G, Sarrigiannis P, Shanmugarajah P, Zis P, Sharrack B, McDermott CJ, Shaw PJ, Hadjivassiliou M. Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations. Neurol Genet. 2018 Oct 24;4(6):e279. doi: 10.1212/NXG.0000000000000279. eCollection 2018 Dec. Erratum In: Neurol Genet. 2018 Dec 03;4(6):e300. Citation on PubMed or Free article on PubMed Central
- Patron M, Sprenger HG, Langer T. m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res. 2018 Mar;28(3):296-306. doi: 10.1038/cr.2018.17. Epub 2018 Feb 16. Citation on PubMed or Free article on PubMed Central
- SPG7 mutations are a common cause of undiagnosed ataxia. Neurology. 2015 May 5;84(18):1911. doi: 10.1212/WNL.0000000000001628. No abstract available. Citation on PubMed or Free article on PubMed Central
- Wilkinson PA, Crosby AH, Turner C, Bradley LJ, Ginsberg L, Wood NW, Schapira AH, Warner TT. A clinical, genetic and biochemical study of SPG7 mutations in hereditary spastic paraplegia. Brain. 2004 May;127(Pt 5):973-80. doi: 10.1093/brain/awh125. Epub 2004 Feb 25. Erratum In: Brain. 2004 Sep;127(Pt 9):2148. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.