

Description
Smith-Magenis syndrome is a developmental disorder that affects behavior, emotions, and learning processes. The major features of this condition include distinctive facial features, sleep disturbances, behavioral problems, mild to moderate intellectual disability, and delayed speech and language skills.
Most people with Smith-Magenis syndrome have a broad, square-shaped face with deep-set eyes, full cheeks, and a prominent lower jaw. The middle of the face and the bridge of the nose often appear flattened. The mouth tends to turn downward with a full, outward-curving upper lip. These facial differences can be subtle in early childhood , but they usually become more distinctive in later childhood and adulthood. Dental abnormalities are also common in affected individuals.
, but they usually become more distinctive in later childhood and adulthood. Dental abnormalities are also common in affected individuals.
Disrupted sleep patterns are characteristic of Smith-Magenis syndrome, and they typically begin early in life. Affected people may have trouble falling asleep at night and awaken several times during the night and early morning. They may be very sleepy during the day.
People with Smith-Magenis syndrome typically have affectionate, engaging personalities, but most also have behavioral problems. These include frequent temper tantrums and outbursts, aggression, anxiety, impulsiveness, and difficulty paying attention. Self-injury, including biting, hitting, head banging, and skin picking, is very common. People with Smith-Magenis syndrome may have other behaviors, such as repetitive self-hugging or compulsively licking their fingers and flipping pages of books and magazines (a behavior known as "lick and flip").
Other signs and symptoms of Smith-Magenis syndrome include short stature, abnormal curvature of the spine (scoliosis ), obesity, and a hoarse voice. Some people with this disorder have ear abnormalities that lead to hearing loss. Affected individuals may have eye abnormalities that cause nearsightedness (myopia
), obesity, and a hoarse voice. Some people with this disorder have ear abnormalities that lead to hearing loss. Affected individuals may have eye abnormalities that cause nearsightedness (myopia ) and other vision problems. Although less common, heart and kidney defects also have been reported in people with Smith-Magenis syndrome.
) and other vision problems. Although less common, heart and kidney defects also have been reported in people with Smith-Magenis syndrome.
Frequency
Smith-Magenis syndrome affects at least 1 in 25,000 individuals worldwide. However, researchers believe that many people with this condition are not diagnosed, so the true prevalence may be closer to 1 in 15,000 individuals.
Causes
In most people with Smith-Magenis syndrome, the condition results from the deletion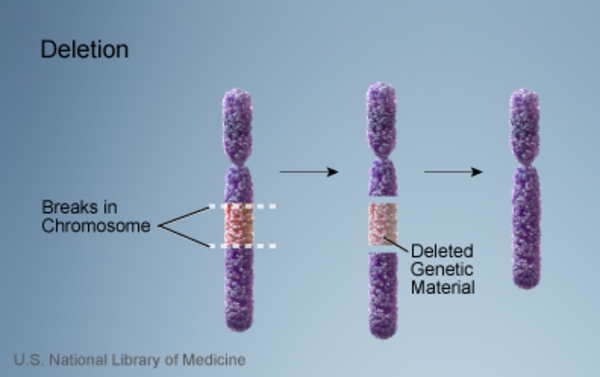 of a small piece of chromosome 17 in each cell. This deletion occurs on the short (p) arm of the chromosome at a position designated p11.2. The deleted segment most often includes approximately 3.7 million DNA building blocks (base pairs), also written as 3.7 megabases (Mb). (An extra copy
of a small piece of chromosome 17 in each cell. This deletion occurs on the short (p) arm of the chromosome at a position designated p11.2. The deleted segment most often includes approximately 3.7 million DNA building blocks (base pairs), also written as 3.7 megabases (Mb). (An extra copy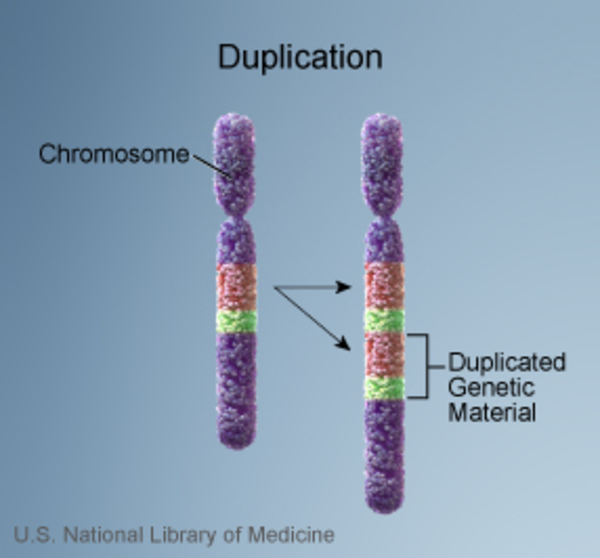 of this segment causes a related condition called Potocki-Lupski syndrome.) Occasionally the deletion is larger or smaller. All of the deletions affect one of the two copies of chromosome 17 in each cell.
of this segment causes a related condition called Potocki-Lupski syndrome.) Occasionally the deletion is larger or smaller. All of the deletions affect one of the two copies of chromosome 17 in each cell.
Although the deleted region contains multiple genes, researchers believe that the loss of one particular gene, RAI1, underlies many of the characteristic features of Smith-Magenis syndrome. All of the deletions known to cause the condition contain this gene. The RAI1 gene provides instructions for making a protein that helps regulate the activity (expression) of other genes. Although most of the genes regulated by the RAI1 protein have not been identified, this protein appears to control the expression of several genes involved in daily (circadian) rhythms, such as the sleep-wake cycle. Studies suggest that the deletion leads to a reduced amount of RAI1 protein in cells, which disrupts the expression of genes that influence circadian rhythms. These changes may account for the sleep disturbances that occur with Smith-Magenis syndrome. It is unclear how a loss of one copy of the RAI1 gene leads to the other physical, mental, and behavioral problems associated with this condition.
A small percentage of people with Smith-Magenis syndrome have a variant in the RAI1 gene instead of a chromosomal deletion. Although these individuals have many of the major features of the condition, they are less likely than people with a deletion to have short stature, hearing loss, and heart or kidney abnormalities. It is likely that, in people with a deletion, the loss of other genes in the deleted region accounts for these additional signs and symptoms; the role of these genes is under study.
Inheritance
Smith-Magenis syndrome is usually not inherited. This condition typically results from a chromosomal deletion or an RAI1 gene variant that occurs during the formation of reproductive cells (eggs or sperm) or in early fetal development. Most people with Smith-Magenis syndrome have no history of the condition in their family.
In a small number of cases, people with Smith-Magenis syndrome have inherited the deletion or variant from an unaffected mother who had the genetic change only in her egg cells. This phenomenon is called germline mosaicism.
Other Names for This Condition
- 17p- syndrome
- 17p11.2 monosomy
- Chromosome 17p deletion syndrome
- Deletion 17p syndrome
- Partial monosomy 17p
- SMS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Carmona-Mora P, Encina CA, Canales CP, Cao L, Molina J, Kairath P, Young JI, Walz K. Functional and cellular characterization of human Retinoic Acid Induced 1 (RAI1) mutations associated with Smith-Magenis Syndrome. BMC Mol Biol. 2010 Aug 25;11:63. doi: 10.1186/1471-2199-11-63. Citation on PubMed or Free article on PubMed Central
- De Leersnyder H. Smith-Magenis syndrome. Handb Clin Neurol. 2013;111:295-6. doi: 10.1016/B978-0-444-52891-9.00034-8. Citation on PubMed
- Elsea SH, Williams SR. Smith-Magenis syndrome: haploinsufficiency of RAI1 results in altered gene regulation in neurological and metabolic pathways. Expert Rev Mol Med. 2011 Apr 19;13:e14. doi: 10.1017/S1462399411001827. Citation on PubMed
- Girirajan S, Elsas LJ 2nd, Devriendt K, Elsea SH. RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J Med Genet. 2005 Nov;42(11):820-8. doi: 10.1136/jmg.2005.031211. Epub 2005 Mar 23. Citation on PubMed or Free article on PubMed Central
- Girirajan S, Vlangos CN, Szomju BB, Edelman E, Trevors CD, Dupuis L, Nezarati M, Bunyan DJ, Elsea SH. Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med. 2006 Jul;8(7):417-27. doi: 10.1097/01.gim.0000228215.32110.89. Citation on PubMed
- Madduri N, Peters SU, Voigt RG, Llorente AM, Lupski JR, Potocki L. Cognitive and adaptive behavior profiles in Smith-Magenis syndrome. J Dev Behav Pediatr. 2006 Jun;27(3):188-92. doi: 10.1097/00004703-200606000-00002. Citation on PubMed
- Neira-Fresneda J, Potocki L. Neurodevelopmental Disorders Associated with Abnormal Gene Dosage: Smith-Magenis and Potocki-Lupski Syndromes. J Pediatr Genet. 2015 Sep;4(3):159-67. doi: 10.1055/s-0035-1564443. Epub 2015 Sep 28. Citation on PubMed or Free article on PubMed Central
- Potocki L, Glaze D, Tan DX, Park SS, Kashork CD, Shaffer LG, Reiter RJ, Lupski JR. Circadian rhythm abnormalities of melatonin in Smith-Magenis syndrome. J Med Genet. 2000 Jun;37(6):428-33. doi: 10.1136/jmg.37.6.428. Citation on PubMed or Free article on PubMed Central
- Smith AC, Dykens E, Greenberg F. Behavioral phenotype of Smith-Magenis syndrome (del 17p11.2). Am J Med Genet. 1998 Mar 28;81(2):179-85. doi: 10.1002/(sici)1096-8628(19980328)81:23.0.co;2-e. Citation on PubMed
- Smith ACM, Boyd KE, Brennan C, Charles J, Elsea SH, Finucane BM, Foster R, Gropman A, Girirajan S, Haas-Givler B. Smith-Magenis Syndrome. 2001 Oct 22 [updated 2022 Mar 10]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1310/ Citation on PubMed
- Tomona N, Smith AC, Guadagnini JP, Hart TC. Craniofacial and dental phenotype of Smith-Magenis syndrome. Am J Med Genet A. 2006 Dec 1;140(23):2556-61. doi: 10.1002/ajmg.a.31371. Citation on PubMed
- Zori RT, Lupski JR, Heju Z, Greenberg F, Killian JM, Gray BA, Driscoll DJ, Patel PI, Zackowski JL. Clinical, cytogenetic, and molecular evidence for an infant with Smith-Magenis syndrome born from a mother having a mosaic 17p11.2p12 deletion. Am J Med Genet. 1993 Sep 15;47(4):504-11. doi: 10.1002/ajmg.1320470414. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.