

Description
SLC4A1-associated distal renal tubular acidosis is a kidney (renal) disorder that sometimes includes blood cell abnormalities. The kidneys normally filter fluid and waste products from the body and remove them in urine; however, in people with distal renal tubular acidosis, the kidneys are unable to remove enough acid from the body, and the blood becomes too acidic. This chemical imbalance is called metabolic acidosis. The inability to remove acids from the body often results in slowed growth and may also lead to softening and weakening of the bones, called rickets in children and osteomalacia in adults. This bone disorder is characterized by bone pain, bowed legs, and difficulty walking. In addition, most children and adults with SLC4A1-associated distal renal tubular acidosis have excess calcium in the urine (hypercalciuria), calcium deposits in the kidneys (nephrocalcinosis), and kidney stones (nephrolithiasis). In rare cases, these kidney abnormalities lead to life-threatening kidney failure. Affected individuals may also have low levels of potassium in the blood (hypokalemia).
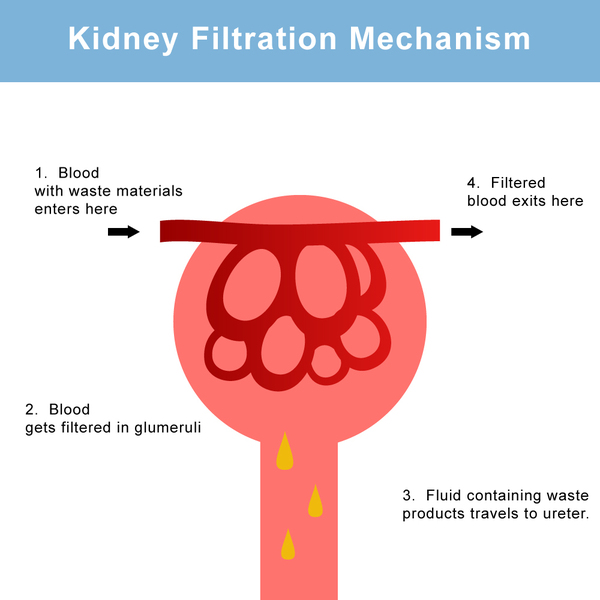
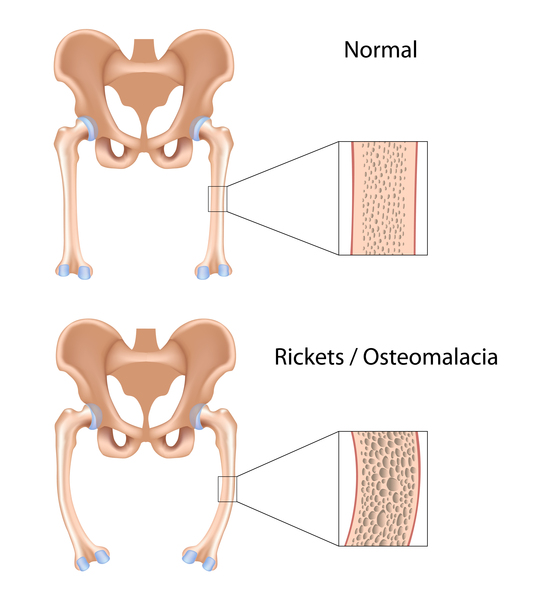
Individuals with the features described above have complete distal renal tubular acidosis, which usually becomes apparent in childhood. Some people do not develop metabolic acidosis even though their kidneys have trouble removing acids; these individuals are said to have incomplete distal renal tubular acidosis. Additionally, these individuals may have other features of distal renal tubular acidosis, such as bone problems and kidney stones. Often, people who initially have incomplete distal renal tubular acidosis develop metabolic acidosis later in life.
Some people with SLC4A1-associated distal renal tubular acidosis also have blood cell abnormalities. These can vary in severity from no symptoms to a condition called hemolytic anemia, in which red blood cells prematurely break down (undergo hemolysis), causing a shortage of red blood cells (anemia). Hemolytic anemia can lead to unusually pale skin (pallor), extreme tiredness (fatigue), shortness of breath (dyspnea), and an enlarged spleen (splenomegaly).

There are two forms of SLC4A1-associated distal renal tubular acidosis; they are distinguished by their inheritance pattern. The autosomal dominant form is more common and is usually less severe than the autosomal recessive form. The autosomal dominant form can be associated with incomplete or complete distal renal tubular acidosis and is rarely associated with blood cell abnormalities. The autosomal recessive form is always associated with complete distal renal tubular acidosis and is more commonly associated with blood cell abnormalities, although not everyone with this form has abnormal blood cells.
Frequency
The prevalence of SLC4A1-associated distal renal tubular acidosis is unknown. The condition is most common in Southeast Asia, especially Thailand.
Causes
Both the autosomal dominant and autosomal recessive forms of SLC4A1-associated distal renal tubular acidosis are caused by mutations in the SLC4A1 gene. This gene provides instructions for making the anion exchanger 1 (AE1) protein, which transports negatively charged atoms (anions) across cell membranes. Specifically, AE1 exchanges negatively charged atoms of chlorine (chloride ions) for negatively charged bicarbonate molecules (bicarbonate ions). The AE1 protein is found in the cell membrane of kidney cells and red blood cells. In kidney cells, the exchange of bicarbonate through AE1 allows acid to be released from the cell into the urine. In red blood cells, AE1 attaches to other proteins that make up the structural framework (the cytoskeleton) of the cells, helping to maintain their structure.
The SLC4A1 gene mutations involved in either form of SLC4A1-associated distal renal tubular acidosis lead to production of altered AE1 proteins that cannot get to the correct location in the cell membrane. In the autosomal dominant form of the condition, gene mutations affect only one copy of the SLC4A1 gene, and normal AE1 protein is produced from the other copy. However, the altered protein attaches to the normal protein and keeps it from getting to the correct location, leading to a severe reduction or absence of AE1 protein in the cell membrane. In autosomal recessive distal renal tubular acidosis, both copies of the SLC4A1 gene are mutated, so all of the protein produced from this gene is altered and not able to get to the correct location. Improper location or absence of AE1 in kidney cell membranes disrupts bicarbonate exchange, and as a result, acid cannot be released into the urine. Instead, the acid builds up in the blood in most affected individuals, leading to metabolic acidosis and the other features of complete distal renal tubular acidosis. It is not clear why some people develop metabolic acidosis and others do not. Researchers suggest that in individuals with incomplete distal renal tubular acidosis, another mechanism is able to help regulate blood acidity (pH) and keep metabolic acidosis from developing.
In red blood cells, interaction with a protein called glycophorin A can often help the altered AE1 protein get to the cell membrane where it can perform its function, which explains why most people with SLC4A1-associated distal renal tubular acidosis do not have blood cell abnormalities. However, some altered AE1 proteins cannot be helped by glycophorin A and are not found in the cell membrane. Without AE1, the red blood cells are unstable; breakdown of these abnormal red blood cells may lead to hemolytic anemia.
Some people have nonhereditary forms of distal renal tubular acidosis; these forms can be caused by immune system problems or other conditions that damage the kidneys. These individuals often have additional signs and symptoms related to the original condition.
Inheritance
SLC4A1-associated distal renal tubular acidosis can have different patterns of inheritance. It is usually inherited in an autosomal dominant pattern, which means one copy of the altered SLC4A1 gene in each cell is sufficient to cause the disorder. In most cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Less commonly, SLC4A1-associated distal renal tubular acidosis has an autosomal recessive pattern of inheritance, which means a mutation must occur in both copies of the SLC4A1 gene for the condition to develop. This pattern occurs with certain types of SLC4A1 gene mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Classic distal renal tubular acidosis
- Renal tubular acidosis type I
- RTA, classic type
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 2009 Jun;212(Pt 11):1672-83. doi: 10.1242/jeb.029454. Citation on PubMed or Free article on PubMed Central
- Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012 Oct;27(10):3691-704. doi: 10.1093/ndt/gfs442. Citation on PubMed
- Cordat E, Kittanakom S, Yenchitsomanus PT, Li J, Du K, Lukacs GL, Reithmeier RA. Dominant and recessive distal renal tubular acidosis mutations of kidney anion exchanger 1 induce distinct trafficking defects in MDCK cells. Traffic. 2006 Feb;7(2):117-28. doi: 10.1111/j.1600-0854.2005.00366.x. Citation on PubMed
- Khositseth S, Sirikanaerat A, Khoprasert S, Opastirakul S, Kingwatanakul P, Thongnoppakhun W, Yenchitsomanus PT. Hematological abnormalities in patients with distal renal tubular acidosis and hemoglobinopathies. Am J Hematol. 2008 Jun;83(6):465-71. doi: 10.1002/ajh.21151. Citation on PubMed
- Ungsupravate D, Sawasdee N, Khositseth S, Udomchaiprasertkul W, Khoprasert S, Li J, Reithmeier RA, Yenchitsomanus PT. Impaired trafficking and intracellular retention of mutant kidney anion exchanger 1 proteins (G701D and A858D) associated with distal renal tubular acidosis. Mol Membr Biol. 2010 Apr;27(2-3):92-103. doi: 10.3109/09687681003588020. Citation on PubMed
- Yenchitsomanus PT, Kittanakom S, Rungroj N, Cordat E, Reithmeier RA. Molecular mechanisms of autosomal dominant and recessive distal renal tubular acidosis caused by SLC4A1 (AE1) mutations. J Mol Genet Med. 2005 Nov 16;1(2):49-62. doi: 10.4172/1747-0862.1000013. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.