

Description
Sialidosis is a severe inherited disorder that affects many organs and tissues, including the nervous system. This disorder is divided into two types, which are distinguished by the age at which symptoms appear and the severity of features.
Sialidosis type I, also referred to as cherry-red spot myoclonus syndrome, is the less severe form of this condition. People with type I develop signs and symptoms of sialidosis in their teens or twenties. Initially, affected individuals experience problems walking (gait disturbance) and/or a loss of sharp vision (reduced visual acuity). Individuals with sialidosis type I also experience muscle twitches (myoclonus), difficulty coordinating movements (ataxia), leg tremors, and seizures. The myoclonus worsens over time, causing difficulty sitting, standing, or walking. People with sialidosis type I eventually require wheelchair assistance. Affected individuals have progressive vision problems, including impaired color vision or night blindness. An eye abnormality called a cherry-red spot, which can be identified with an eye examination, is characteristic of this disorder. Sialidosis type I does not affect intelligence or life expectancy.
Sialidosis type II, the more severe type of the disorder, is further divided into congenital, infantile, and juvenile forms. The features of congenital sialidosis type II can develop before birth. This form of sialidosis is associated with an abnormal buildup of fluid in the abdominal cavity (ascites) or widespread swelling before birth caused by fluid accumulation (hydrops fetalis). Affected infants may also have an enlarged liver and spleen (hepatosplenomegaly), abnormal bone development (dysostosis multiplex), and distinctive facial features that are often described as "coarse." As a result of these serious health problems, individuals with congenital sialidosis type II usually are stillborn or die soon after birth.
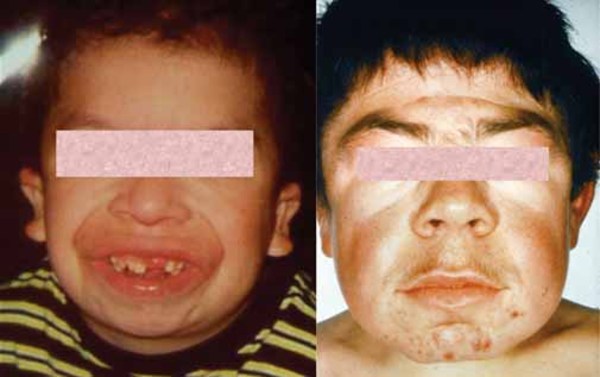
Infantile sialidosis type II shares some features with the congenital form, although the signs and symptoms are slightly less severe and begin within the first year of life. Features of the infantile form include hepatosplenomegaly, dysostosis multiplex, "coarse" facial features, short stature, and intellectual disability. As children with infantile sialidosis type II get older, they may develop myoclonus and cherry-red spots. Other signs and symptoms include hearing loss, overgrowth of the gums (gingival hyperplasia), and widely spaced teeth. Affected individuals may survive into childhood or adolescence.
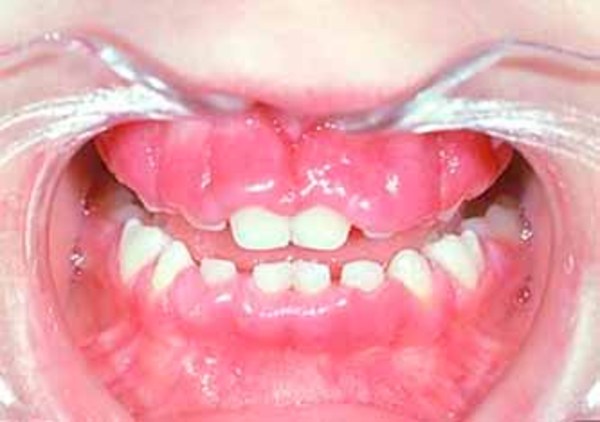
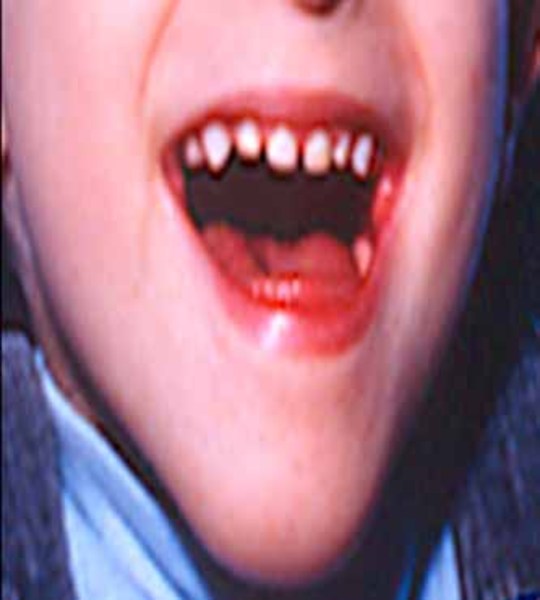
The juvenile form has the least severe signs and symptoms of the different forms of sialidosis type II. Features of this condition usually appear in late childhood and may include mildly "coarse" facial features, mild bone abnormalities, cherry-red spots, myoclonus, intellectual disability, and dark red spots on the skin (angiokeratomas). The life expectancy of individuals with juvenile sialidosis type II varies depending on the severity of symptoms.
Frequency
The overall prevalence of sialidosis is unknown. Sialidosis type I appears to be more common in people with Italian ancestry.
Causes
Mutations in the NEU1 gene cause sialidosis. This gene provides instructions for making an enzyme called neuraminidase 1 (NEU1), which is found in lysosomes. Lysosomes are compartments within the cell that use enzymes to digest and recycle materials. The NEU1 enzyme helps break down large sugar molecules attached to certain proteins by removing a substance known as sialic acid.

Mutations in the NEU1 gene lead to a shortage (deficiency) of the NEU1 enzyme. When this enzyme is lacking, sialic acid-containing compounds accumulate inside lysosomes. Conditions such as sialidosis that cause molecules to build up inside lysosomes are called lysosomal storage disorders. People with sialidosis type II have mutations that severely reduce or eliminate NEU1 enzyme activity. Individuals with sialidosis type I have mutations that result in some functional NEU1 enzyme. It is unclear exactly how the accumulation of large molecules within lysosomes leads to the signs and symptoms of sialidosis.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Cherry red spot myoclonus syndrome
- Mucolipidosis I
- Mucolipidosis type I
- Myoclonus cherry red spot syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Caciotti A, Di Rocco M, Filocamo M, Grossi S, Traverso F, d'Azzo A, Cavicchi C, Messeri A, Guerrini R, Zammarchi E, Donati MA, Morrone A. Type II sialidosis: review of the clinical spectrum and identification of a new splicing defect with chitotriosidase assessment in two patients. J Neurol. 2009 Nov;256(11):1911-5. doi: 10.1007/s00415-009-5213-4. Epub 2009 Jul 1. Citation on PubMed
- Lai SC, Chen RS, Wu Chou YH, Chang HC, Kao LY, Huang YZ, Weng YH, Chen JK, Hwu WL, Lu CS. A longitudinal study of Taiwanese sialidosis type 1: an insight into the concept of cherry-red spot myoclonus syndrome. Eur J Neurol. 2009 Aug;16(8):912-9. doi: 10.1111/j.1468-1331.2009.02622.x. Epub 2009 Apr 14. Citation on PubMed
- Pattison S, Pankarican M, Rupar CA, Graham FL, Igdoura SA. Five novel mutations in the lysosomal sialidase gene (NEU1) in type II sialidosis patients and assessment of their impact on enzyme activity and intracellular targeting using adenovirus-mediated expression. Hum Mutat. 2004 Jan;23(1):32-9. doi: 10.1002/humu.10278. Citation on PubMed
- Seyrantepe V, Poupetova H, Froissart R, Zabot MT, Maire I, Pshezhetsky AV. Molecular pathology of NEU1 gene in sialidosis. Hum Mutat. 2003 Nov;22(5):343-52. doi: 10.1002/humu.10268. Citation on PubMed
- Wu X, Steigelman KA, Bonten E, Hu H, He W, Ren T, Zuo J, d'Azzo A. Vacuolization and alterations of lysosomal membrane proteins in cochlear marginal cells contribute to hearing loss in neuraminidase 1-deficient mice. Biochim Biophys Acta. 2010 Feb;1802(2):259-68. doi: 10.1016/j.bbadis.2009.10.008. Epub 2009 Oct 24. Citation on PubMed or Free article on PubMed Central
- Yogalingam G, Bonten EJ, van de Vlekkert D, Hu H, Moshiach S, Connell SA, d'Azzo A. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev Cell. 2008 Jul;15(1):74-86. doi: 10.1016/j.devcel.2008.05.005. Citation on PubMed or Free article on PubMed Central
- Zanoteli E, van de Vlekkert D, Bonten EJ, Hu H, Mann L, Gomero EM, Harris AJ, Ghersi G, d'Azzo A. Muscle degeneration in neuraminidase 1-deficient mice results from infiltration of the muscle fibers by expanded connective tissue. Biochim Biophys Acta. 2010 Jul-Aug;1802(7-8):659-72. doi: 10.1016/j.bbadis.2010.04.002. Epub 2010 Apr 11. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.