

Description
Saethre-Chotzen syndrome is a genetic condition characterized by the premature fusion of certain skull bones (craniosynostosis). This early fusion prevents the skull from growing normally and affects the shape of the head and face.
Most people with Saethre-Chotzen syndrome have prematurely fused skull bones along the coronal suture, the growth line that goes over the head from ear to ear. Other parts of the skull may be malformed as well. These changes can result in an abnormally shaped head, a high forehead, a low frontal hairline, droopy eyelids (ptosis), widely spaced eyes, and a broad nasal bridge. One side of the face may appear noticeably different from the other (facial asymmetry). Most people with Saethre-Chotzen syndrome also have small, rounded ears.

The signs and symptoms of Saethre-Chotzen syndrome vary widely, even among affected individuals in the same family. This condition can cause mild changes in the hands and feet, such as partial fusion of the skin between the second and third fingers on each hand and a broad or duplicated first (big) toe. Delayed development and learning difficulties have been reported, although most people with this condition are of normal intelligence. Less common signs and symptoms of Saethre-Chotzen syndrome include short stature, abnormalities of the bones of the spine (the vertebra), hearing loss, and heart defects.
Robinow-Sorauf syndrome is a condition with features similar to those of Saethre-Chotzen syndrome, including craniosynostosis and broad or duplicated great toes. It was once considered a separate disorder, but was found to result from mutations in the same gene and is now thought to be a variant of Saethre-Chotzen syndrome.
Frequency
Saethre-Chotzen syndrome has an estimated prevalence of 1 in 50,000 people.
Causes
Mutations in the TWIST1 gene cause Saethre-Chotzen syndrome. The TWIST1 gene provides instructions for making a protein that plays an important role in early development. This protein is a transcription factor, which means that it attaches (binds) to specific regions of DNA and helps control the activity of particular genes. The TWIST1 protein is active in cells that give rise to bones, muscles, and other tissues in the head and face. It is also involved in the development of the limbs.
Mutations in the TWIST1 gene prevent one copy of the gene in each cell from making any functional protein. A shortage of the TWIST1 protein affects the development and maturation of cells in the skull, face, and limbs. These abnormalities underlie the signs and symptoms of Saethre-Chotzen syndrome, including the premature fusion of certain skull bones.
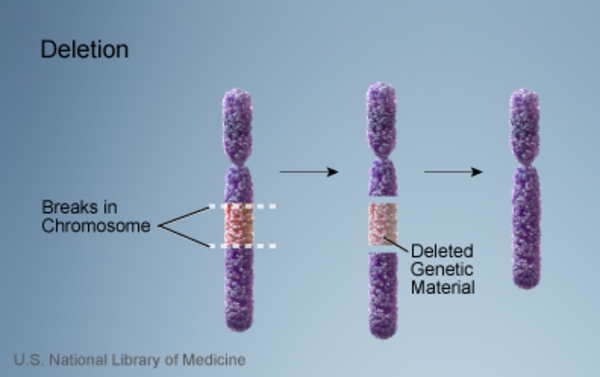
In a small number of people with Saethre-Chotzen syndrome, the condition is caused by a structural chromosomal abnormality, such as a deletion or rearrangement of genetic material, in the region of chromosome 7 that contains the TWIST1 gene. When Saethre-Chotzen syndrome is caused by a chromosomal deletion instead of a mutation within the TWIST1 gene, affected children are much more likely to have intellectual disability, developmental delay, and learning difficulties. These features are typically not seen in classic cases of Saethre-Chotzen syndrome. Researchers believe that a loss of other genes on chromosome 7 may be responsible for these additional features.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases may result from new mutations in the gene. These cases occur in people with no history of the disorder in their family.


Some people with a TWIST1 mutation do not have any of the obvious features of Saethre-Chotzen syndrome. These people are still at risk of passing on the gene mutation and may have a child with craniosynostosis and the other typical signs and symptoms of the condition.
Other Names for This Condition
- Acrocephalosyndactyly III
- Acrocephalosyndactyly, type III
- Acrocephaly, skull asymmetry, and mild syndactyly
- ACS III
- ACS3
- Chotzen syndrome
- Dysostosis craniofacialis with hypertelorism
- SCS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Cai J, Goodman BK, Patel AS, Mulliken JB, Van Maldergem L, Hoganson GE, Paznekas WA, Ben-Neriah Z, Sheffer R, Cunningham ML, Daentl DL, Jabs EW. Increased risk for developmental delay in Saethre-Chotzen syndrome is associated with TWIST deletions: an improved strategy for TWIST mutation screening. Hum Genet. 2003 Dec;114(1):68-76. doi: 10.1007/s00439-003-1012-7. Epub 2003 Sep 25. Citation on PubMed
- Cai J, Shoo BA, Sorauf T, Jabs EW. A novel mutation in the TWIST gene, implicated in Saethre-Chotzen syndrome, is found in the original case of Robinow-Sorauf syndrome. Clin Genet. 2003 Jul;64(1):79-82. doi: 10.1034/j.1399-0004.2003.00098.x. No abstract available. Citation on PubMed
- Chun K, Teebi AS, Jung JH, Kennedy S, Laframboise R, Meschino WS, Nakabayashi K, Scherer SW, Ray PN, Teshima I. Genetic analysis of patients with the Saethre-Chotzen phenotype. Am J Med Genet. 2002 Jun 15;110(2):136-43. doi: 10.1002/ajmg.10400. Citation on PubMed
- Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res. 2007 May;10(2):67-81. doi: 10.1111/j.1601-6343.2007.00389.x. Citation on PubMed
- de Heer IM, de Klein A, van den Ouweland AM, Vermeij-Keers C, Wouters CH, Vaandrager JM, Hovius SE, Hoogeboom JM. Clinical and genetic analysis of patients with Saethre-Chotzen syndrome. Plast Reconstr Surg. 2005 Jun;115(7):1894-902; discussion 1903-5. doi: 10.1097/01.prs.0000165278.72168.51. Citation on PubMed
- Gallagher ER, Ratisoontorn C, Cunningham ML. Saethre-Chotzen Syndrome. 2003 May 16 [updated 2019 Jan 24]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1189/ Citation on PubMed
- Gripp KW, Zackai EH, Stolle CA. Mutations in the human TWIST gene. Hum Mutat. 2000;15(2):150-5. doi: 10.1002/(SICI)1098-1004(200002)15:23.0.CO;2-D. Erratum In: Hum Mutat 2000;15(5):479. Citation on PubMed
- Kress W, Schropp C, Lieb G, Petersen B, Busse-Ratzka M, Kunz J, Reinhart E, Schafer WD, Sold J, Hoppe F, Pahnke J, Trusen A, Sorensen N, Krauss J, Collmann H. Saethre-Chotzen syndrome caused by TWIST 1 gene mutations: functional differentiation from Muenke coronal synostosis syndrome. Eur J Hum Genet. 2006 Jan;14(1):39-48. doi: 10.1038/sj.ejhg.5201507. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.