

Description
Rothmund-Thomson syndrome is a rare condition that affects many parts of the body, especially the skin. People with this condition typically develop redness on the cheeks between ages 3 months and 6 months. Over time the rash spreads to the arms and legs, causing patchy changes in skin coloring, areas of thinning skin (atrophy), and small clusters of blood vessels just under the skin (telangiectases). These skin problems persist for life and are collectively known as poikiloderma.
Rothmund-Thomson syndrome is also characterized by sparse hair, eyebrows, and eyelashes; slow growth and small stature; abnormalities of the teeth and nails; and gastrointestinal problems in infancy, such as chronic diarrhea and vomiting. Some affected children develop a clouding of the lens of the eye (cataract), which affects vision. Many people with this disorder have skeletal abnormalities including absent or malformed bones, fused bones, and low bone mineral density (osteopenia or osteoporosis). Some of these abnormalities affect the development of bones in the forearms and the thumbs, and are known as radial ray malformations.

People with Rothmund-Thomson syndrome have an increased risk of developing cancer, particularly a form of bone cancer called osteosarcoma. These bone tumors most often develop during childhood or adolescence. Several types of skin cancer, including basal cell carcinoma and squamous cell carcinoma, are also more common in people with this disorder.
The varied signs and symptoms of Rothmund-Thomson syndrome overlap with features of other disorders, namely Baller-Gerold syndrome and RAPADILINO syndrome. These syndromes are also characterized by radial ray defects, skeletal abnormalities, and slow growth. All of these conditions can be caused by mutations in the same gene. Based on these similarities, researchers are investigating whether Rothmund-Thomson syndrome, Baller-Gerold syndrome, and RAPADILINO syndrome are separate disorders or part of a single syndrome with overlapping signs and symptoms.
Frequency
Rothmund-Thomson syndrome is a rare disorder; its incidence is unknown. About 300 people with this condition have been reported worldwide in scientific studies.
Causes
Mutations in the RECQL4 gene cause about two-thirds of all cases of Rothmund-Thomson syndrome. This gene provides instructions for making one member of a protein family called RecQ helicases. Helicases are enzymes that bind to DNA and temporarily unwind the two spiral strands (double helix) of the DNA molecule. This unwinding is necessary for copying (replicating) DNA in preparation for cell division, and for repairing damaged DNA. The RECQL4 protein helps stabilize genetic information in the body's cells and plays a role in replicating and repairing DNA.
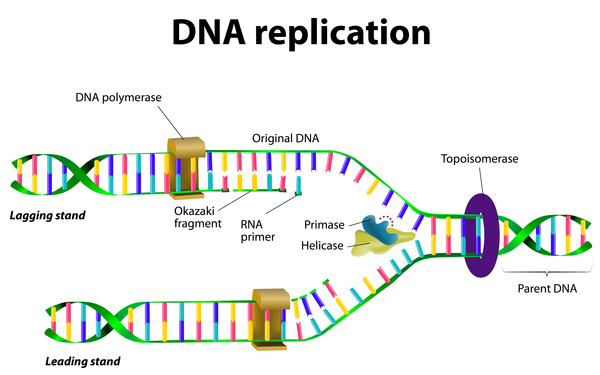
RECQL4 mutations lead to the production of an abnormally short, nonfunctional version of the RECQL4 protein or prevent cells from making any of this protein. A shortage of the RECQL4 protein may prevent normal DNA replication and repair, causing widespread damage to a person's genetic information over time. It is unclear how a loss of this protein's activity leads to the specific features of Rothmund-Thomson syndrome.
In about one-third of individuals with Rothmund-Thomson syndrome, no mutation in the RECQL4 gene has been found. The cause of the condition in these individuals is unknown; however, researchers suspect that these cases may result from mutations in a gene related to the RECQL4 gene.
In some cases, chromosomal abnormalities have been identified in people with Rothmund-Thomson syndrome. These abnormalities include extra or missing genetic material, usually from chromosome 7 or chromosome 8, in some of an affected person's cells. Researchers believe that these chromosomal changes arise because of the overall instability of an affected person's genetic information; they do not cause the disorder.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Congenital poikiloderma
- Poikiloderma atrophicans and cataract
- Poikiloderma congenitale
- Poikiloderma congenitale of Rothmund-Thomson
- RTS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Hicks MJ, Roth JR, Kozinetz CA, Wang LL. Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol. 2007 Feb 1;25(4):370-5. doi: 10.1200/JCO.2006.08.4558. Citation on PubMed
- Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006 Jan 28;232(1):107-20. doi: 10.1016/j.canlet.2005.07.042. Epub 2005 Nov 3. Citation on PubMed
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010 Jan 29;5:2. doi: 10.1186/1750-1172-5-2. Citation on PubMed or Free article on PubMed Central
- Miozzo M, Castorina P, Riva P, Dalpra L, Fuhrman Conti AM, Volpi L, Hoe TS, Khoo A, Wiegant J, Rosenberg C, Larizza L. Chromosomal instability in fibroblasts and mesenchymal tumors from 2 sibs with Rothmund-Thomson syndrome. Int J Cancer. 1998 Aug 12;77(4):504-10. doi: 10.1002/(sici)1097-0215(19980812)77:43.0.co;2-y. Citation on PubMed
- Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, Crandall B, Hannula-Jouppi K, Hennekam R, Herzog D, Keymolen K, Lipsanen-Nyman M, Miny P, Plon SE, Riedl S, Sarkar A, Vargas FR, Verloes A, Wang LL, Kaariainen H, Kestila M. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009 Feb;17(2):151-8. doi: 10.1038/ejhg.2008.154. Epub 2008 Aug 20. Citation on PubMed or Free article on PubMed Central
- Wang LL, Gannavarapu A, Kozinetz CA, Levy ML, Lewis RA, Chintagumpala MM, Ruiz-Maldanado R, Contreras-Ruiz J, Cunniff C, Erickson RP, Lev D, Rogers M, Zackai EH, Plon SE. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003 May 7;95(9):669-74. doi: 10.1093/jnci/95.9.669. Citation on PubMed
- Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, Plon SE. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet. 2001 Jul 22;102(1):11-7. doi: 10.1002/1096-8628(20010722)102:13.0.co;2-a. Citation on PubMed
- Wang LL, Plon SE. Rothmund-Thomson Syndrome. 1999 Oct 6 [updated 2020 Jun 4]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1237/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.