

Description
Roberts syndrome is a genetic disorder characterized by limb and facial abnormalities. Affected individuals also grow slowly before and after birth. Mild to severe intellectual impairment occurs in about half of all people with Roberts syndrome.
Children with Roberts syndrome are born with abnormalities of all four limbs. They have shortened arm and leg bones (hypomelia), particularly the bones in their forearms and lower legs. In severe cases, the limbs may be so short that the hands and feet are located very close to the body (phocomelia). People with Roberts syndrome may also have abnormal or missing fingers and toes, and joint deformities (contractures) commonly occur at the elbows and knees. The limb abnormalities are very similar on the right and left sides of the body, but arms are usually more severely affected than legs.
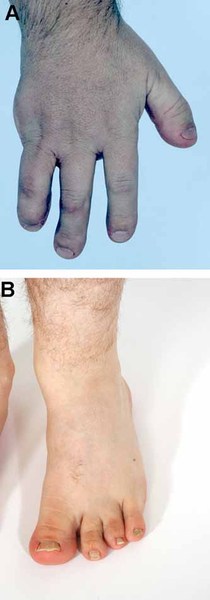
Individuals with Roberts syndrome typically have numerous facial abnormalities, including an opening in the lip (a cleft lip) with or without an opening in the roof of the mouth (cleft palate), a small chin (micrognathia), ear abnormalities, wide-set eyes (hypertelorism), outer corners of the eyes that point downward (down-slanting palpebral fissures), small nostrils, and a beaked nose. They may have a small head size (microcephaly) or clouding of the clear front covering of the eyes (corneal opacities). In severe cases affected individuals have a sac-like protrusion of the brain (encephalocele) at the front of their head. In addition, people with Roberts syndrome may have heart, kidney, and genital abnormalities.
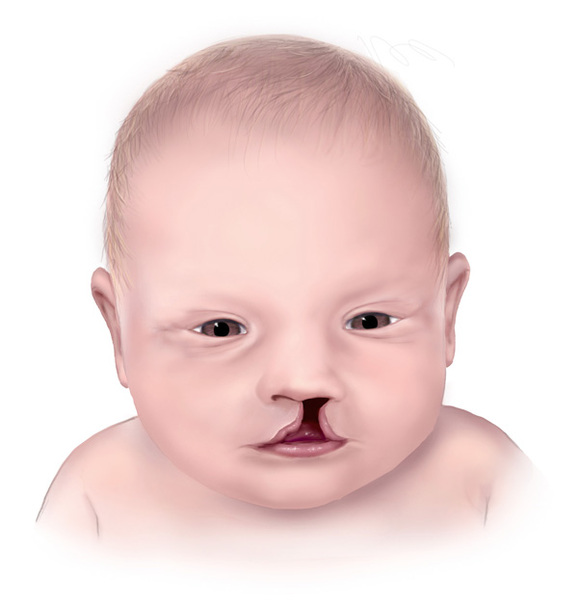

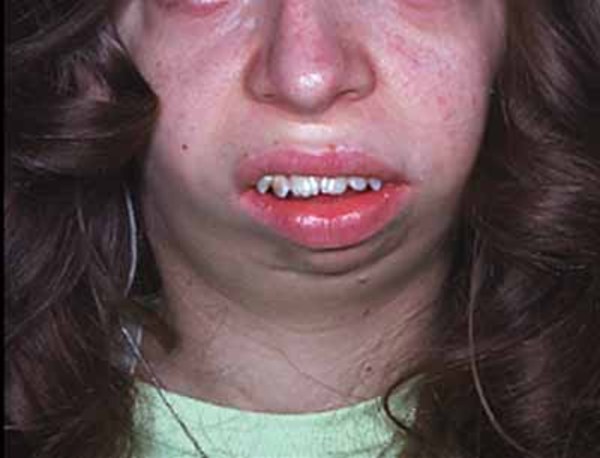

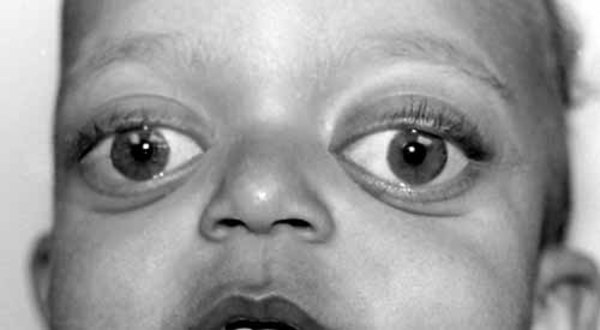

Infants with a severe form of Roberts syndrome are often stillborn or die shortly after birth. Mildly affected individuals may live into adulthood. A condition called SC phocomelia syndrome was originally thought to be distinct from Roberts syndrome; however, it is now considered to be a mild variant. "SC" represents the first letters of the surnames of the two families first diagnosed with this disorder.
Frequency
Roberts syndrome is a rare disorder. Its prevalence is unknown.
Causes
Mutations in the ESCO2 gene cause Roberts syndrome. This gene provides instructions for making a protein that is important for proper chromosome separation during cell division. Before cells divide, they must copy all of their chromosomes. The copied DNA from each chromosome is arranged into two identical structures, called sister chromatids. The ESCO2 protein plays an important role in establishing the glue that holds the sister chromatids together until the chromosomes are ready to separate.
All identified mutations in the ESCO2 gene prevent the cell from producing any functional ESCO2 protein, which causes some of the glue between sister chromatids to be missing around the chromosome's constriction point (centromere). In Roberts syndrome, cells respond to abnormal sister chromatid attachment by delaying cell division. Delayed cell division can be a signal that the cell should undergo self-destruction. The signs and symptoms of Roberts syndrome may result from the loss of cells from various tissues during early development. Because both mildly and severely affected individuals lack any functional ESCO2 protein, the underlying cause of the variation in disease severity remains unknown. Researchers suspect that other genetic and environmental factors may be involved.

Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Appelt-Gerken-Lenz syndrome
- Hypomelia hypotrichosis facial hemangioma syndrome
- Pseudothalidomide syndrome
- RBS
- Roberts-SC phocomelia syndrome
- SC phocomelia syndrome
- SC pseudothalidomide syndrome
- SC syndrome
- Tetraphocomelia-cleft palate syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dorsett D. Roles of the sister chromatid cohesion apparatus in gene expression, development, and human syndromes. Chromosoma. 2007 Feb;116(1):1-13. doi: 10.1007/s00412-006-0072-6. Epub 2006 Jul 4. Citation on PubMed or Free article on PubMed Central
- Gordillo M, Vega H, Trainer AH, Hou F, Sakai N, Luque R, Kayserili H, Basaran S, Skovby F, Hennekam RC, Uzielli ML, Schnur RE, Manouvrier S, Chang S, Blair E, Hurst JA, Forzano F, Meins M, Simola KO, Raas-Rothschild A, Schultz RA, McDaniel LD, Ozono K, Inui K, Zou H, Jabs EW. The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum Mol Genet. 2008 Jul 15;17(14):2172-80. doi: 10.1093/hmg/ddn116. Epub 2008 Apr 14. Citation on PubMed
- Hou F, Zou H. Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion. Mol Biol Cell. 2005 Aug;16(8):3908-18. doi: 10.1091/mbc.e04-12-1063. Epub 2005 Jun 15. Citation on PubMed or Free article on PubMed Central
- McNairn AJ, Gerton JL. Cohesinopathies: One ring, many obligations. Mutat Res. 2008 Dec 1;647(1-2):103-11. doi: 10.1016/j.mrfmmm.2008.08.010. Epub 2008 Aug 22. Citation on PubMed
- Resta N, Susca FC, Di Giacomo MC, Stella A, Bukvic N, Bagnulo R, Simone C, Guanti G. A homozygous frameshift mutation in the ESCO2 gene: evidence of intertissue and interindividual variation in Nmd efficiency. J Cell Physiol. 2006 Oct;209(1):67-73. doi: 10.1002/jcp.20708. Citation on PubMed
- Schule B, Oviedo A, Johnston K, Pai S, Francke U. Inactivating mutations in ESCO2 cause SC phocomelia and Roberts syndrome: no phenotype-genotype correlation. Am J Hum Genet. 2005 Dec;77(6):1117-28. doi: 10.1086/498695. Epub 2005 Oct 31. Citation on PubMed or Free article on PubMed Central
- Vega H, Gordillo M, Jabs EW. ESCO2 Spectrum Disorder. 2006 Apr 18 [updated 2020 Mar 26]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1153/ Citation on PubMed
- Vega H, Trainer AH, Gordillo M, Crosier M, Kayserili H, Skovby F, Uzielli ML, Schnur RE, Manouvrier S, Blair E, Hurst JA, Forzano F, Meins M, Simola KO, Raas-Rothschild A, Hennekam RC, Jabs EW. Phenotypic variability in 49 cases of ESCO2 mutations, including novel missense and codon deletion in the acetyltransferase domain, correlates with ESCO2 expression and establishes the clinical criteria for Roberts syndrome. J Med Genet. 2010 Jan;47(1):30-7. doi: 10.1136/jmg.2009.068395. Epub 2009 Jul 1. Citation on PubMed
- Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, Yamada M, van Gosliga D, Kayserili H, Xu C, Ozono K, Jabs EW, Inui K, Joenje H. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet. 2005 May;37(5):468-70. doi: 10.1038/ng1548. Epub 2005 Apr 10. Citation on PubMed
- Whelan G, Kreidl E, Wutz G, Egner A, Peters JM, Eichele G. Cohesin acetyltransferase Esco2 is a cell viability factor and is required for cohesion in pericentric heterochromatin. EMBO J. 2012 Jan 4;31(1):71-82. doi: 10.1038/emboj.2011.381. Epub 2011 Nov 18. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.