

Description
Rett syndrome is a brain disorder that occurs almost exclusively in girls. The most common form of the condition is known as classic Rett syndrome. After birth, girls with classic Rett syndrome have 6 to 18 months of apparently normal development before developing severe problems with language and communication, learning, coordination, and other brain functions. Early in childhood, affected girls lose purposeful use of their hands and begin making repeated hand wringing, washing, or clapping motions. They tend to grow more slowly than other children and about three-quarters have a small head size (microcephaly). Other signs and symptoms that can develop include breathing abnormalities, spitting or drooling, unusual eye movements such as intense staring or excessive blinking, cold hands and feet, irritability, sleep disturbances, seizures, and an abnormal side-to-side curvature of the spine (scoliosis).


Researchers have described several variant or atypical forms of Rett syndrome, which can be milder or more severe than the classic form.
Rett syndrome is part of a spectrum of disorders with the same genetic cause. Other disorders on the spectrum include PPM-X syndrome, MECP2 duplication syndrome, and MECP2-related severe neonatal encephalopathy. These other conditions can affect males.
Frequency
This condition affects an estimated 1 in 9,000 to 10,000 females.
Causes
Mutations in a gene called MECP2 underlie almost all cases of classic Rett syndrome and some variant forms of the condition. This gene provides instructions for making a protein (MeCP2) that is critical for normal brain function. Although the exact function of the MeCP2 protein is unclear, it is likely involved in maintaining connections (synapses) between nerve cells (neurons). It may also be necessary for the normal function of other types of brain cells.

The MeCP2 protein is thought to help regulate the activity of genes in the brain. This protein may also control the production of different versions of certain proteins in brain cells. Mutations in the MECP2 gene alter the MeCP2 protein or result in the production of less protein, which appears to disrupt the normal function of neurons and other cells in the brain. Specifically, studies suggest that changes in the MeCP2 protein may reduce the activity of certain neurons and impair their ability to communicate with one another. It is unclear how these changes lead to the specific features of Rett syndrome.
Several conditions with signs and symptoms overlapping those of Rett syndrome have been found to result from mutations in other genes. These conditions, including FOXG1 syndrome and CDKL5 deficiency disorder, were previously thought to be variant forms of Rett syndrome. However, doctors and researchers have identified some important differences between the conditions, so they are now usually considered to be separate disorders.
Inheritance
In more than 99 percent of people with Rett syndrome, there is no history of the disorder in their family. Many of these cases result from new mutations in the MECP2 gene.
A few families with more than one affected family member have been described. These cases helped researchers determine that classic Rett syndrome and variants caused by MECP2 gene mutations have an X-linked dominant pattern of inheritance. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. The inheritance is dominant if one copy of the altered gene in each cell is sufficient to cause the condition.
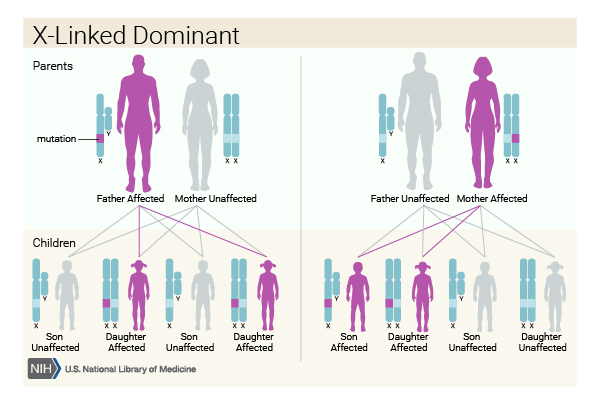
Males with mutations in the MECP2 gene often die in infancy. However, a small number of males with a genetic change involving MECP2 have developed signs and symptoms similar to those of Rett syndrome, including intellectual disability, seizures, and movement problems. In males, this condition is described as MECP2-related severe neonatal encephalopathy. The signs and symptoms in some males with an MECP2 gene mutation are on the milder end of the spectrum.
Other Names for This Condition
- Autism-dementia-ataxia-loss of purposeful hand use syndrome
- Rett disorder
- Rett's disorder
- Rett's syndrome
- RTT
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007 Nov 8;56(3):422-37. doi: 10.1016/j.neuron.2007.10.001. Citation on PubMed
- Ehrhart F, Coort SL, Cirillo E, Smeets E, Evelo CT, Curfs LM. Rett syndrome - biological pathways leading from MECP2 to disorder phenotypes. Orphanet J Rare Dis. 2016 Nov 25;11(1):158. doi: 10.1186/s13023-016-0545-5. Citation on PubMed or Free article on PubMed Central
- Gold WA, Krishnarajy R, Ellaway C, Christodoulou J. Rett Syndrome: A Genetic Update and Clinical Review Focusing on Comorbidities. ACS Chem Neurosci. 2018 Feb 21;9(2):167-176. doi: 10.1021/acschemneuro.7b00346. Epub 2017 Dec 15. Citation on PubMed
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey ME, Schanen NC, Zappella M, Renieri A, Huppke P, Percy AK; RettSearch Consortium. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010 Dec;68(6):944-50. doi: 10.1002/ana.22124. Citation on PubMed or Free article on PubMed Central
- Neul JL, Zoghbi HY. Rett syndrome: a prototypical neurodevelopmental disorder. Neuroscientist. 2004 Apr;10(2):118-28. doi: 10.1177/1073858403260995. Citation on PubMed
- Percy AK, Lane JB. Rett syndrome: model of neurodevelopmental disorders. J Child Neurol. 2005 Sep;20(9):718-21. doi: 10.1177/08830738050200090301. Citation on PubMed
- Samaco RC, Neul JL. Complexities of Rett syndrome and MeCP2. J Neurosci. 2011 Jun 1;31(22):7951-9. doi: 10.1523/JNEUROSCI.0169-11.2011. No abstract available. Citation on PubMed or Free article on PubMed Central
- Zoghbi HY. Rett syndrome: what do we know for sure? Nat Neurosci. 2009 Mar;12(3):239-40. doi: 10.1038/nn0309-239. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.