

Description
Renpenning syndrome is a disorder that almost exclusively affects males, causing developmental delay, moderate to severe intellectual disability, and distinctive physical features. Individuals with Renpenning syndrome typically have short stature and a small head size (microcephaly). Facial features characteristic of this disorder include a long, narrow face; outside corners of the eyes that point upward (upslanting palpebral fissures); a long, bulbous nose with a low-hanging separation between the nostrils (overhanging columella); a shortened space between the nose and mouth (philtrum); and cup-shaped ears. Males with Renpenning syndrome generally have small testes. Seizures and wasting away (atrophy) of muscles used for movement (skeletal muscles) may also occur in this disorder.

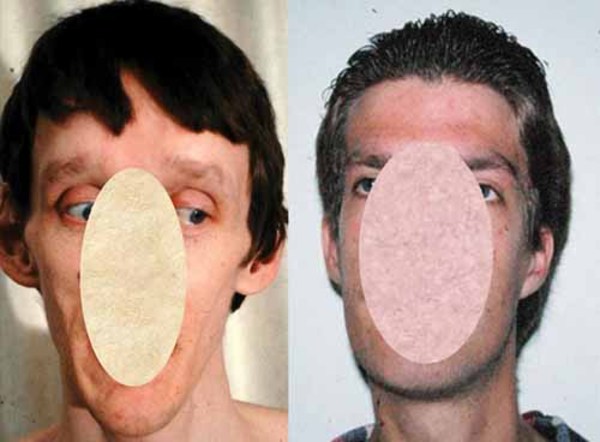
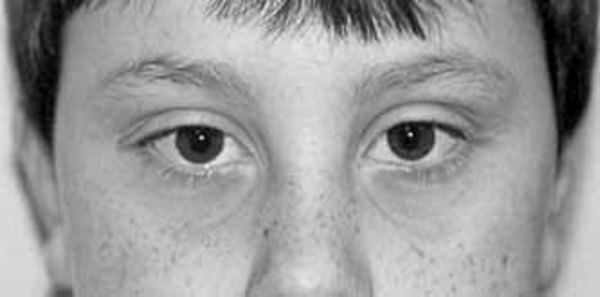
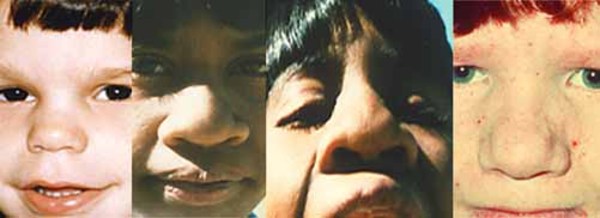
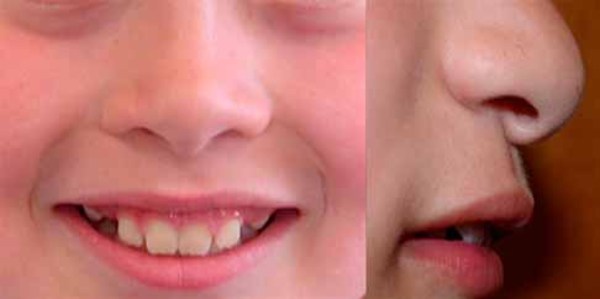

About 20 percent of individuals with Renpenning syndrome also have other features, which may include a gap or split in structures that make up the eye (coloboma), an opening in the roof of the mouth (cleft palate), heart abnormalities, or malformations of the anus.

Certain combinations of the features that often occur in Renpenning syndrome are sometimes called by other names, such as Golabi-Ito-Hall syndrome or Sutherland-Haan syndrome. However, all these syndromes, which have the same genetic cause, are now generally grouped under the term Renpenning syndrome.
Frequency
Renpenning syndrome is a rare disorder; its prevalence is unknown. More than 60 affected individuals in at least 15 families have been identified.
Causes
Renpenning syndrome is caused by mutations in the PQBP1 gene. This gene provides instructions for making a protein called polyglutamine-binding protein 1. This protein attaches (binds) to stretches of multiple copies of a protein building block (amino acid) called glutamine in certain other proteins.

While the specific function of polyglutamine-binding protein 1 is not well understood, it is believed to play a role in processing and transporting RNA, a chemical cousin of DNA that serves as the genetic blueprint for the production of proteins.
In nerve cells (neurons) such as those in the brain, polyglutamine-binding protein 1 is found in structures called RNA granules. These granules allow the transport and storage of RNA within the cell. The RNA is held within the granules until the genetic information it carries is translated to produce proteins or until cellular signals or environmental factors trigger the RNA to be degraded. Through these mechanisms, polyglutamine-binding protein 1 is thought to help control the way genetic information is used (gene expression) in neurons. This control is important for normal brain development.

Most of the mutations in the PQBP1 gene that cause Renpenning syndrome result in an abnormally short polyglutamine-binding protein 1. The function of a shortened or otherwise abnormal protein is likely impaired and interferes with normal gene expression in neurons, resulting in abnormal development of the brain and the signs and symptoms of Renpenning syndrome.
Inheritance
This condition is inherited in an X-linked recessive pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation typically has to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

Other Names for This Condition
- Golabi-Ito-Hall syndrome
- Hamel cerebropalatocardiac syndrome
- Porteous syndrome
- Sutherland-Haan syndrome
- X-linked intellectual deficit due to PQBP1 mutations
- X-linked intellectual deficit, Renpenning type
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Cossee M, Demeer B, Blanchet P, Echenne B, Singh D, Hagens O, Antin M, Finck S, Vallee L, Dollfus H, Hegde S, Springell K, Thelma BK, Woods G, Kalscheuer V, Mandel JL. Exonic microdeletions in the X-linked PQBP1 gene in mentally retarded patients: a pathogenic mutation and in-frame deletions of uncertain effect. Eur J Hum Genet. 2006 Apr;14(4):418-25. doi: 10.1038/sj.ejhg.5201593. Citation on PubMed
- Flynn M, Zou YS, Milunsky A. Whole gene duplication of the PQBP1 gene in syndrome resembling Renpenning. Am J Med Genet A. 2011 Jan;155A(1):141-4. doi: 10.1002/ajmg.a.33756. Epub 2010 Dec 9. Citation on PubMed
- Germanaud D, Rossi M, Bussy G, Gerard D, Hertz-Pannier L, Blanchet P, Dollfus H, Giuliano F, Bennouna-Greene V, Sarda P, Sigaudy S, Curie A, Vincent MC, Touraine R, des Portes V. The Renpenning syndrome spectrum: new clinical insights supported by 13 new PQBP1-mutated males. Clin Genet. 2011 Mar;79(3):225-35. doi: 10.1111/j.1399-0004.2010.01551.x. Epub 2010 Oct 18. Citation on PubMed
- Kleefstra T, Franken CE, Arens YH, Ramakers GJ, Yntema HG, Sistermans EA, Hulsmans CF, Nillesen WN, van Bokhoven H, de Vries BB, Hamel BC. Genotype-phenotype studies in three families with mutations in the polyglutamine-binding protein 1 gene (PQBP1). Clin Genet. 2004 Oct;66(4):318-26. doi: 10.1111/j.1399-0004.2004.00308.x. Citation on PubMed
- Kunde SA, Musante L, Grimme A, Fischer U, Muller E, Wanker EE, Kalscheuer VM. The X-chromosome-linked intellectual disability protein PQBP1 is a component of neuronal RNA granules and regulates the appearance of stress granules. Hum Mol Genet. 2011 Dec 15;20(24):4916-31. doi: 10.1093/hmg/ddr430. Epub 2011 Sep 20. Citation on PubMed
- Lenski C, Abidi F, Meindl A, Gibson A, Platzer M, Frank Kooy R, Lubs HA, Stevenson RE, Ramser J, Schwartz CE. Novel truncating mutations in the polyglutamine tract binding protein 1 gene (PQBP1) cause Renpenning syndrome and X-linked mental retardation in another family with microcephaly. Am J Hum Genet. 2004 Apr;74(4):777-80. doi: 10.1086/383205. No abstract available. Citation on PubMed or Free article on PubMed Central
- Lubs H, Abidi FE, Echeverri R, Holloway L, Meindl A, Stevenson RE, Schwartz CE. Golabi-Ito-Hall syndrome results from a missense mutation in the WW domain of the PQBP1 gene. J Med Genet. 2006 Jun;43(6):e30. doi: 10.1136/jmg.2005.037556. Citation on PubMed or Free article on PubMed Central
- Martinez-Garay I, Tomas M, Oltra S, Ramser J, Molto MD, Prieto F, Meindl A, Kutsche K, Martinez F. A two base pair deletion in the PQBP1 gene is associated with microphthalmia, microcephaly, and mental retardation. Eur J Hum Genet. 2007 Jan;15(1):29-34. doi: 10.1038/sj.ejhg.5201717. Epub 2006 Oct 11. Citation on PubMed
- Musante L, Kunde SA, Sulistio TO, Fischer U, Grimme A, Frints SG, Schwartz CE, Martinez F, Romano C, Ropers HH, Kalscheuer VM. Common pathological mutations in PQBP1 induce nonsense-mediated mRNA decay and enhance exclusion of the mutant exon. Hum Mutat. 2010 Jan;31(1):90-8. doi: 10.1002/humu.21146. Citation on PubMed
- Stevenson RE, Bennett CW, Abidi F, Kleefstra T, Porteous M, Simensen RJ, Lubs HA, Hamel BC, Schwartz CE. Renpenning syndrome comes into focus. Am J Med Genet A. 2005 May 1;134(4):415-21. doi: 10.1002/ajmg.a.30664. Citation on PubMed
- Tapia VE, Nicolaescu E, McDonald CB, Musi V, Oka T, Inayoshi Y, Satteson AC, Mazack V, Humbert J, Gaffney CJ, Beullens M, Schwartz CE, Landgraf C, Volkmer R, Pastore A, Farooq A, Bollen M, Sudol M. Y65C missense mutation in the WW domain of the Golabi-Ito-Hall syndrome protein PQBP1 affects its binding activity and deregulates pre-mRNA splicing. J Biol Chem. 2010 Jun 18;285(25):19391-401. doi: 10.1074/jbc.M109.084525. Epub 2010 Apr 21. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.