

Description
Pyruvate dehydrogenase deficiency is characterized by the buildup of a chemical called lactic acid in the body and a variety of neurological problems. Signs and symptoms of this condition usually first appear shortly after birth, and they can vary widely among affected individuals. The most common feature is a potentially life-threatening buildup of lactic acid (lactic acidosis), which can cause nausea, vomiting, severe breathing problems, and an abnormal heartbeat. People with pyruvate dehydrogenase deficiency usually have neurological problems as well. Most have delayed development of mental abilities and motor skills such as sitting and walking. Other neurological problems can include intellectual disability, seizures, weak muscle tone (hypotonia), poor coordination, and difficulty walking. Some affected individuals have abnormal brain structures, such as underdevelopment of the tissue connecting the left and right halves of the brain (corpus callosum), wasting away (atrophy) of the exterior part of the brain known as the cerebral cortex, or patches of damaged tissue (lesions) on some parts of the brain. Because of the severe health effects, many individuals with pyruvate dehydrogenase deficiency do not survive past childhood, although some may live into adolescence or adulthood.
Frequency
Pyruvate dehydrogenase deficiency is believed to be a rare condition; however, its prevalence is unknown.
Causes
The genes involved in pyruvate dehydrogenase deficiency each provide instructions for making a protein that is a component of a group of proteins called the pyruvate dehydrogenase complex. This complex plays an important role in the pathways that convert the energy from food into a form that cells can use. The pyruvate dehydrogenase complex converts a molecule called pyruvate, which is formed from the breakdown of carbohydrates, into another molecule called acetyl-CoA. This conversion is essential to begin the series of chemical reactions that produce energy for cells.
The pyruvate dehydrogenase complex is made up of multiple copies of several enzymes called E1, E2, and E3, each of which performs part of the chemical reaction that converts pyruvate to acetyl-CoA. In addition, other proteins included in the complex ensure its proper function. One of these proteins, E3 binding protein, attaches E3 to the complex and provides the correct structure for the complex to perform its function. Other associated proteins control the activity of the complex: pyruvate dehydrogenase phosphatase turns on (activates) the complex, while pyruvate dehydrogenase kinase turns off (inhibits) the complex.
The E1 enzyme, also called pyruvate dehydrogenase, is composed of four parts (subunits): two alpha subunits (called E1 alpha) and two beta subunits (called E1 beta). Mutations in the gene that provides instructions for making E1 alpha, the PDHA1 gene, are the most common cause of pyruvate dehydrogenase deficiency, accounting for approximately 80 percent of cases. These mutations lead to a shortage of E1 alpha protein or result in an abnormal protein that cannot function properly. A decrease in functional E1 alpha leads to reduced activity of the pyruvate dehydrogenase complex.
Other components of the pyruvate dehydrogenase complex are also involved in pyruvate dehydrogenase deficiency. Mutations in the genes that provide instructions for E1 beta (the PDHB gene), the E2 enzyme (the DLAT gene), E3 binding protein (the PDHX gene), and pyruvate dehydrogenase phosphatase (the PDP1 gene) have been identified in people with this condition. Although it is unclear how mutations in each of these genes affect the complex, reduced functioning of one component of the complex appears to impair the activity of the whole complex. As with PDHA1 gene mutations, changes in these other genes lead to a reduction of pyruvate dehydrogenase complex activity.
With decreased function of this complex, pyruvate builds up and is converted in another chemical reaction to lactic acid. The excess lactic acid causes lactic acidosis in affected individuals. In addition, the production of cellular energy is diminished. The brain, which requires especially large amounts of energy, is severely affected, resulting in the neurological problems associated with pyruvate dehydrogenase deficiency.
Inheritance
Pyruvate dehydrogenase deficiency can have different inheritance patterns. When the condition is caused by mutations in the PDHA1 gene, it is inherited in an X-linked pattern. The PDHA1 gene is located on the X chromosome, which is one of the two sex chromosomes. In males, who have only one X chromosome, a mutation in the only copy of the gene in each cell is sufficient to cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

In females, who have two copies of the X chromosome, one altered copy of the PDHA1 gene in each cell can lead to less severe features of the condition or may cause no signs or symptoms at all. However, many females with one altered copy of this gene have pyruvate dehydrogenase deficiency similar to affected males because the X chromosome with the normal copy of the PDHA1 gene is turned off through a process called X-inactivation. Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
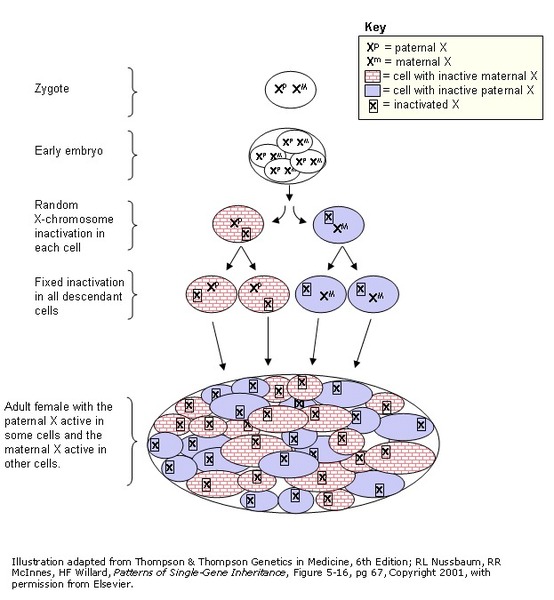
Research shows that females with pyruvate dehydrogenase deficiency caused by mutation of the PDHA1 gene often have skewed X-inactivation, which results in the inactivation of the X chromosome with the normal copy of the PDHA1 gene in most cells of the body. This skewed X-inactivation causes the chromosome with the mutated PDHA1 gene to be expressed in more than half of cells.
When caused by mutations in other genes, pyruvate dehydrogenase deficiency is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Ataxia with lactic acidosis
- Intermittent ataxia with pyruvate dehydrogenase deficiency
- PDH deficiency
- PDHC deficiency
- Pyruvate dehydrogenase complex deficiency
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Pyruvate dehydrogenase complex deficiency

- Genetic Testing Registry: Pyruvate dehydrogenase E1-alpha deficiency
- Genetic Testing Registry: Pyruvate dehydrogenase E1-beta deficiency
- Genetic Testing Registry: Pyruvate dehydrogenase E2 deficiency
- Genetic Testing Registry: Pyruvate dehydrogenase E3-binding protein deficiency
- Genetic Testing Registry: Pyruvate dehydrogenase phosphatase deficiency
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Brown RM, Head RA, Brown GK. Pyruvate dehydrogenase E3 binding protein deficiency. Hum Genet. 2002 Feb;110(2):187-91. doi: 10.1007/s00439-001-0665-3. Epub 2002 Jan 22. Citation on PubMed
- Chun K, MacKay N, Petrova-Benedict R, Federico A, Fois A, Cole DE, Robertson E, Robinson BH. Mutations in the X-linked E1 alpha subunit of pyruvate dehydrogenase: exon skipping, insertion of duplicate sequence, and missense mutations leading to the deficiency of the pyruvate dehydrogenase complex. Am J Hum Genet. 1995 Mar;56(3):558-69. Citation on PubMed or Free article on PubMed Central
- Ganetzky R, McCormick EM, Falk MJ. Primary Pyruvate Dehydrogenase Complex Deficiency Overview. 2021 Jun 17. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK571223/ Citation on PubMed
- Head RA, Brown RM, Zolkipli Z, Shahdadpuri R, King MD, Clayton PT, Brown GK. Clinical and genetic spectrum of pyruvate dehydrogenase deficiency: dihydrolipoamide acetyltransferase (E2) deficiency. Ann Neurol. 2005 Aug;58(2):234-41. doi: 10.1002/ana.20550. Citation on PubMed
- Hong YS, Kerr DS, Liu TC, Lusk M, Powell BR, Patel MS. Deficiency of dihydrolipoamide dehydrogenase due to two mutant alleles (E340K and G101del). Analysis of a family and prenatal testing. Biochim Biophys Acta. 1997 Dec 31;1362(2-3):160-8. doi: 10.1016/s0925-4439(97)00073-2. Citation on PubMed
- Imbard A, Boutron A, Vequaud C, Zater M, de Lonlay P, de Baulny HO, Barnerias C, Mine M, Marsac C, Saudubray JM, Brivet M. Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural implications of novel amino acid substitutions in E1 protein. Mol Genet Metab. 2011 Dec;104(4):507-16. doi: 10.1016/j.ymgme.2011.08.008. Epub 2011 Aug 18. Citation on PubMed
- Maj MC, MacKay N, Levandovskiy V, Addis J, Baumgartner ER, Baumgartner MR, Robinson BH, Cameron JM. Pyruvate dehydrogenase phosphatase deficiency: identification of the first mutation in two brothers and restoration of activity by protein complementation. J Clin Endocrinol Metab. 2005 Jul;90(7):4101-7. doi: 10.1210/jc.2005-0123. Epub 2005 Apr 26. Citation on PubMed
- Okajima K, Korotchkina LG, Prasad C, Rupar T, Phillips JA 3rd, Ficicioglu C, Hertecant J, Patel MS, Kerr DS. Mutations of the E1beta subunit gene (PDHB) in four families with pyruvate dehydrogenase deficiency. Mol Genet Metab. 2008 Apr;93(4):371-80. doi: 10.1016/j.ymgme.2007.10.135. Epub 2008 Mar 4. Citation on PubMed
- Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012 Jan;105(1):34-43. doi: 10.1016/j.ymgme.2011.09.032. Epub 2011 Oct 7. Erratum In: Mol Genet Metab. 2012 Jul;106(3):384. Citation on PubMed or Free article on PubMed Central
- Willemsen M, Rodenburg RJ, Teszas A, van den Heuvel L, Kosztolanyi G, Morava E. Females with PDHA1 gene mutations: a diagnostic challenge. Mitochondrion. 2006 Jun;6(3):155-9. doi: 10.1016/j.mito.2006.03.001. Epub 2006 May 19. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.