

Description
Primary hyperoxaluria is a rare condition characterized by recurrent kidney and bladder stones. The condition often results in end stage renal disease (ESRD), which is a life-threatening condition that prevents the kidneys from filtering fluids and waste products from the body effectively.
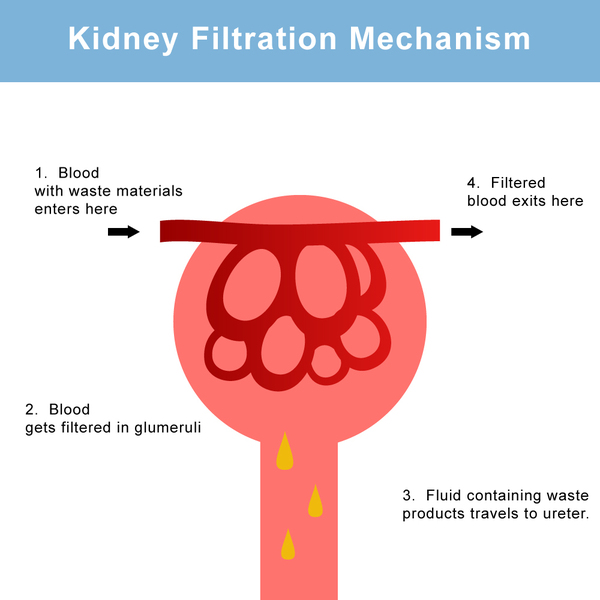
Primary hyperoxaluria results from the overproduction of a substance called oxalate. Oxalate is filtered through the kidneys and excreted as a waste product in urine, leading to abnormally high levels of this substance in urine (hyperoxaluria). During its excretion, oxalate can combine with calcium to form calcium oxalate, a hard compound that is the main component of kidney and bladder stones. Deposits of calcium oxalate can damage the kidneys and other organs and lead to blood in the urine (hematuria), urinary tract infections, kidney damage, ESRD, and injury to other organs. Over time, kidney function decreases such that the kidneys can no longer excrete as much oxalate as they receive. As a result oxalate levels in the blood rise, and the substance gets deposited in tissues throughout the body (systemic oxalosis), particularly in bones and the walls of blood vessels. Oxalosis in bones can cause fractures.
There are three types of primary hyperoxaluria that differ in their severity and genetic cause. In primary hyperoxaluria type 1, kidney stones typically begin to appear anytime from childhood to early adulthood, and ESRD can develop at any age. Primary hyperoxaluria type 2 is similar to type 1, but ESRD develops later in life. In primary hyperoxaluria type 3, affected individuals often develop kidney stones in early childhood, but few cases of this type have been described so additional signs and symptoms of this type are unclear.
Frequency
Primary hyperoxaluria is estimated to affect 1 in 58,000 individuals worldwide. Type 1 is the most common form, accounting for approximately 80 percent of cases. Types 2 and 3 each account for about 10 percent of cases.
Causes
Mutations in the AGXT, GRHPR, and HOGA1 genes cause primary hyperoxaluria types 1, 2, and 3, respectively. These genes provide instructions for making enzymes that are involved in the breakdown and processing of protein building blocks (amino acids) and other compounds. The enzyme produced from the HOGA1 gene is involved in the breakdown of an amino acid, which results in the formation of a compound called glyoxylate. This compound is further broken down by the enzymes produced from the AGXT and GRHPR genes.

Mutations in the AGXT, GRHPR, or HOGA1 gene lead to a decrease in production or activity of the respective proteins, which prevents the normal breakdown of glyoxylate. AGXT and GRHPR gene mutations result in an accumulation of glyoxylate, which is then converted to oxalate for removal from the body as a waste product. HOGA1 gene mutations also result in excess oxalate, although researchers are unsure as to how this occurs. Oxalate that is not excreted from the body combines with calcium to form calcium oxalate deposits, which can damage the kidneys and other organs.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Congenital oxaluria
- D-glycerate dehydrogenase deficiency
- Glyceric aciduria
- Glycolic aciduria
- Hepatic AGT deficiency
- Hyperoxaluria, primary
- Oxalosis
- Oxaluria, primary
- Peroxisomal alanine:glyoxylate aminotransferase deficiency
- Primary oxalosis
- Primary oxaluria
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Allard L, Cochat P, Leclerc AL, Cachat F, Fichtner C, De Souza VC, Garcia CD, Camoin-Schweitzer MC, Macher MA, Acquaviva-Bourdain C, Bacchetta J. Renal function can be impaired in children with primary hyperoxaluria type 3. Pediatr Nephrol. 2015 Oct;30(10):1807-13. doi: 10.1007/s00467-015-3090-x. Epub 2015 May 14. Citation on PubMed
- Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013 Aug 15;369(7):649-58. doi: 10.1056/NEJMra1301564. No abstract available. Erratum In: N Engl J Med. 2013 Nov 28;369(22):2168. Citation on PubMed
- Hopp K, Cogal AG, Bergstralh EJ, Seide BM, Olson JB, Meek AM, Lieske JC, Milliner DS, Harris PC; Rare Kidney Stone Consortium. Phenotype-Genotype Correlations and Estimated Carrier Frequencies of Primary Hyperoxaluria. J Am Soc Nephrol. 2015 Oct;26(10):2559-70. doi: 10.1681/ASN.2014070698. Epub 2015 Feb 2. Citation on PubMed or Free article on PubMed Central
- Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012 Jun 12;8(8):467-75. doi: 10.1038/nrneph.2012.113. Citation on PubMed
- Milliner DS, Harris PC, Sas DJ, Cogal AG, Lieske JC. Primary Hyperoxaluria Type 1. 2002 Jun 19 [updated 2022 Feb 10]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1283/ Citation on PubMed
- Milliner DS, Harris PC, Sas DJ, Lieske JC. Primary Hyperoxaluria Type 3. 2015 Sep 24 [updated 2023 Feb 9]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK316514/ Citation on PubMed
- Rumsby G, Hulton SA. Primary Hyperoxaluria Type 2. 2008 Dec 2 [updated 2017 Dec 21]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK2692/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.