

Description
PGM3-congenital disorder of glycosylation (PGM3-CDG) is an inherited condition that primarily affects the immune system but can also involve other areas of the body. The pattern and severity of this disorder's signs and symptoms typically vary.
Most people with PGM3-CDG have impaired immune function (immune deficiency). Many have a shortage of white blood cells (leukopenia), which normally protect the body from infection. Because affected individuals lack the necessary immune cells to fight off certain bacteria, viruses, and fungi, they are prone to repeated and persistent infections that often occur in the lungs, ears, skin, or gastrointestinal tract. In severe cases of PGM3-CDG, impaired bone marrow function may lead to a decrease in the production of all blood cells, resulting in a condition called bone marrow failure. Affected individuals usually also have allergies, asthma, or an inflammatory skin condition called eczema. People with PGM3-CDG may develop autoimmunity, which occurs when the body attacks its own tissues and organs by mistake. Persistent illness may cause affected children to grow more slowly than other individuals.
Additionally, people with PGM3-CDG often have abnormally high levels of immune system proteins called antibodies (also known as immunoglobulins), particularly immunoglobulin E (IgE). Antibodies help protect the body against infection by attaching to specific foreign particles and germs, marking them for destruction. The effect of abnormal levels of antibodies in PGM3-CDG is unclear.
People with PGM3-CDG often have intellectual disability, delayed development, and weak muscle tone (hypotonia). Many affected individuals have skeletal abnormalities involving the ribs or bones in the hands, feet, or spine. Some people with this condition have distinct facial features, such as a flat or sunken appearance of the middle of the face (midface hypoplasia), small chin (micrognathia), full lips, downturned corners of the mouth, and wide nostrils that open to the front rather than downward. PGM3-CDG can also cause problems in the lungs, gastrointestinal tract, and kidneys.
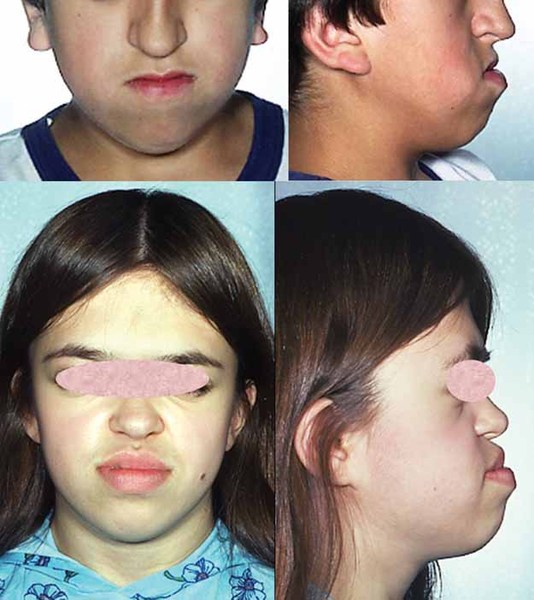
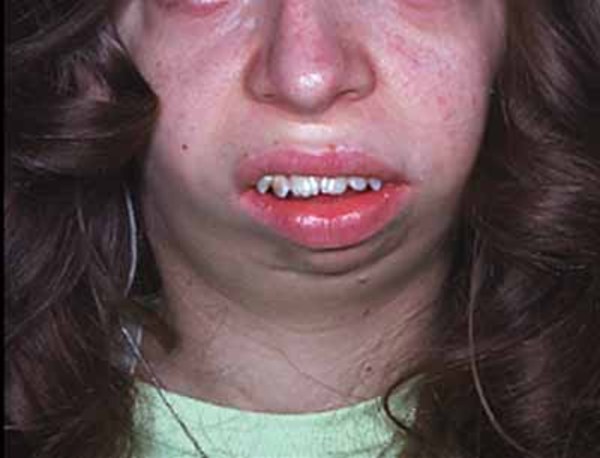
Lifespan varies widely in people with PGM3-CDG; some do not survive past infancy while others live into late adulthood.
Frequency
PGM3-CDG is a rare disorder, although its prevalence is unknown. Approximately 40 people with the condition have been described worldwide.
Causes
Mutations in the PGM3 gene cause PGM3-CDG. This gene provides instructions for making an enzyme called phosphoglucomutase 3 (PGM3). The PGM3 enzyme is involved in a process called glycosylation, which attaches groups of sugar molecules (oligosaccharides) to proteins. During this process, complex chains of sugar molecules (oligosaccharides) are added to proteins and fats (lipids). Glycosylation modifies proteins and lipids so they can perform a wider variety of functions.
Mutations in the PGM3 gene lead to the production of a PGM3 enzyme with reduced activity. Without a properly functioning enzyme, glycosylation cannot proceed normally. The wide variety of signs and symptoms in PGM3-CDG are likely due to impaired glycosylation of proteins and lipids that are needed for the normal function of many organs and tissues. Immune system proteins are highly dependent on glycosylation to function normally, which likely explains why people with PGM3-CDG have immune deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- AGM1 deficiency
- CID due to PGM3 deficiency
- Combined immunodeficiency due to PGM3 deficiency
- Deficiency of N-acetylglucosamine-phosphate mutase 1
- Deficiency of phosphoglucomutase 3
- Immunodeficiency 23
- Immunodeficiency with hyper IgE and cognitive impairment
- Immunodeficiency-vasculitis-myoclonus syndrome
- PGM3 deficiency
- PGM3-CDG
- PGM3-related congenital disorder of glycosylation
- Phosphoglucomutase 3 deficiency
- Phosphoglucomutase deficiency type 3
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Jaeken J, Lefeber DJ, Matthijs G. Clinical Utility Gene Card for: PGM3 defective congenital disorder of glycosylation. Eur J Hum Genet. 2019 Nov;27(11):1757-1760. doi: 10.1038/s41431-019-0453-y. Epub 2019 Jun 23. Citation on PubMed
- Lundin KE, Hamasy A, Backe PH, Moens LN, Falk-Sorqvist E, Elgstoen KB, Morkrid L, Bjoras M, Granert C, Norlin AC, Nilsson M, Christensson B, Stenmark S, Smith CI. Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene. Clin Immunol. 2015 Dec;161(2):366-72. doi: 10.1016/j.clim.2015.10.002. Epub 2015 Oct 19. Citation on PubMed or Free article on PubMed Central
- Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, Patiroglu T, Unal E, Ozdemir MA, Jouhadi Z, Khadir K, Ben-Khemis L, Ben-Ali M, Ben-Mustapha I, Borchani L, Pfeifer D, Jakob T, Khemiri M, Asplund AC, Gustafsson MO, Lundin KE, Falk-Sorqvist E, Moens LN, Gungor HE, Engelhardt KR, Dziadzio M, Stauss H, Fleckenstein B, Meier R, Prayitno K, Maul-Pavicic A, Schaffer S, Rakhmanov M, Henneke P, Kraus H, Eibel H, Kolsch U, Nadifi S, Nilsson M, Bejaoui M, Schaffer AA, Smith CI, Dell A, Barbouche MR, Grimbacher B. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol. 2014 May;133(5):1410-9, 1419.e1-13. doi: 10.1016/j.jaci.2014.02.025. Epub 2014 Apr 1. Citation on PubMed or Free article on PubMed Central
- Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, Erichsen HC, Gambin T, Elgstoen KB, Bjoras M, Wlodarski MW, Kruger M, Jhangiani SN, Muzny DM, Patel A, Raymond KM, Sasa GS, Krance RA, Martinez CA, Abraham SM, Speckmann C, Ehl S, Hall P, Forbes LR, Merckoll E, Westvik J, Nishimura G, Rustad CF, Abrahamsen TG, Ronnestad A, Osnes LT, Egeland T, Rodningen OK, Beck CR; Baylor-Johns Hopkins Center for Mendelian Genomics; Boerwinkle EA, Gibbs RA, Lupski JR, Orange JS, Lausch E, Hanson IC. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. 2014 Jul 3;95(1):96-107. doi: 10.1016/j.ajhg.2014.05.007. Epub 2014 Jun 12. Citation on PubMed or Free article on PubMed Central
- Yang L, Fliegauf M, Grimbacher B. Hyper-IgE syndromes: reviewing PGM3 deficiency. Curr Opin Pediatr. 2014 Dec;26(6):697-703. doi: 10.1097/MOP.0000000000000158. Citation on PubMed
- Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, Jing H, Kim ES, Biancalana M, Wolfe LA, DiMaggio T, Matthews HF, Kranick SM, Stone KD, Holland SM, Reich DS, Hughes JD, Mehmet H, McElwee J, Freeman AF, Freeze HH, Su HC, Milner JD. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014 May;133(5):1400-9, 1409.e1-5. doi: 10.1016/j.jaci.2014.02.013. Epub 2014 Feb 28. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.