

Description
Otospondylomegaepiphyseal dysplasia (OSMED) is a condition characterized by skeletal abnormalities, distinctive facial features, and severe hearing loss. The term "otospondylomegaepiphyseal" refers to the parts of the body that this condition affects: the ears (oto-), the bones of the spine (spondylo-), and the ends (epiphyses) of long bones in the arms and legs. The features of this condition significantly overlap those of two similar conditions, Weissenbacher-Zweymüller syndrome and Stickler syndrome type III. All of these conditions are caused by mutations in the same gene, and in some cases, it can be difficult to tell the conditions apart. Some researchers believe they represent a single disorder with a range of signs and symptoms.

People with OSMED are often shorter than average because the long bones in their legs are unusually short. Other skeletal features include enlarged joints; short arms, hands, and fingers; and flattened bones of the spine (platyspondyly). People with the disorder often experience back and joint pain, limited joint movement, and arthritis that begins early in life.

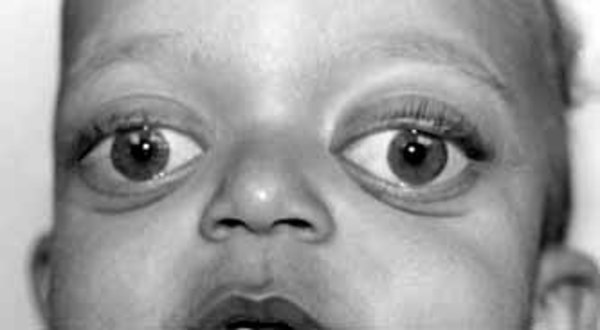
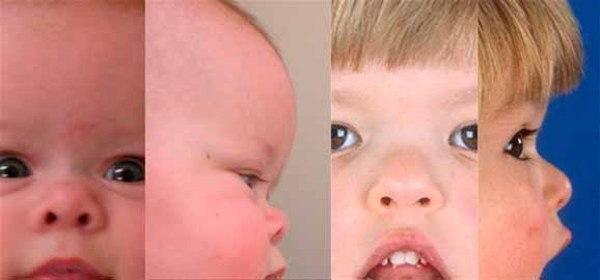
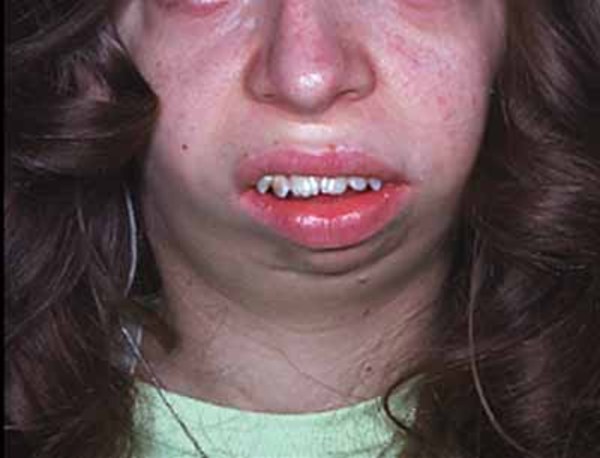
Severe high-frequency hearing loss is common in people with OSMED. Typical facial features include protruding eyes; a flattened bridge of the nose; an upturned nose with a large, rounded tip; and a small lower jaw. Almost all affected infants are born with an opening in the roof of the mouth (a cleft palate).

Frequency
This condition is rare; its prevalence is unknown. Only a few families with OSMED worldwide have been described in the medical literature.
Causes
OSMED is caused by mutations in the COL11A2 gene. This gene provides instructions for making one component of type XI collagen, which is a complex molecule that gives structure and strength to the connective tissues that support the body's joints and organs. Type XI collagen is found in cartilage, a tough but flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone, except for the cartilage that continues to cover and protect the ends of bones and is present in the nose and external ears. Type XI collagen is also part of the inner ear and the nucleus pulposus, which is the center portion of the discs between vertebrae.
The COL11A2 gene mutations that cause OSMED disrupt the production or assembly of type XI collagen molecules. The defective collagen weakens connective tissues in many parts of the body, including the long bones, spine, and inner ears, which impairs bone development and underlies the other signs and symptoms of this condition.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Chondrodystrophy with sensorineural deafness
- Insley-Astley syndrome
- Mega-epiphyseal dwarfism
- Nance-Insley syndrome
- Nance-Sweeney chondrodysplasia
- OSMED
- Oto-spondylo-megaepiphyseal dysplasia
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Harel T, Rabinowitz R, Hendler N, Galil A, Flusser H, Chemke J, Gradstein L, Lifshitz T, Ofir R, Elbedour K, Birk OS. COL11A2 mutation associated with autosomal recessive Weissenbacher-Zweymuller syndrome: molecular and clinical overlap with otospondylomegaepiphyseal dysplasia (OSMED). Am J Med Genet A. 2005 Jan 1;132A(1):33-5. doi: 10.1002/ajmg.a.30371. Citation on PubMed
- Melkoniemi M, Brunner HG, Manouvrier S, Hennekam R, Superti-Furga A, Kaariainen H, Pauli RM, van Essen T, Warman ML, Bonaventure J, Miny P, Ala-Kokko L. Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene. Am J Hum Genet. 2000 Feb;66(2):368-77. doi: 10.1086/302750. Citation on PubMed or Free article on PubMed Central
- Temtamy SA, Mannikko M, Abdel-Salam GM, Hassan NA, Ala-Kokko L, Afifi HH. Oto-spondylo-megaepiphyseal dysplasia (OSMED): clinical and radiological findings in sibs homozygous for premature stop codon mutation in the COL11A2 gene. Am J Med Genet A. 2006 Jun 1;140(11):1189-95. doi: 10.1002/ajmg.a.31205. Citation on PubMed
- van Steensel MA, Buma P, de Waal Malefijt MC, van den Hoogen FH, Brunner HG. Oto- spondylo-megaepiphyseal dysplasia (OSMED): clinical description of three patients homozygous for a missense mutation in the COL11A2 gene. Am J Med Genet. 1997 Jun 13;70(3):315-23. doi: 10.1002/(sici)1096-8628(19970613)70:33.3.co;2-y. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.