

Description
Opitz G/BBB syndrome is a genetic condition that causes several abnormalities along the midline of the body. "G/BBB" represents the first letters of the last names of the families first diagnosed with this disorder and "Opitz" is the last name of the doctor who first described the signs and symptoms. There are two forms of Opitz G/BBB syndrome, X-linked Opitz G/BBB syndrome and autosomal dominant Opitz G/BBB syndrome. The two forms are distinguished by their genetic causes and patterns of inheritance. The signs and symptoms of the two forms are generally the same.
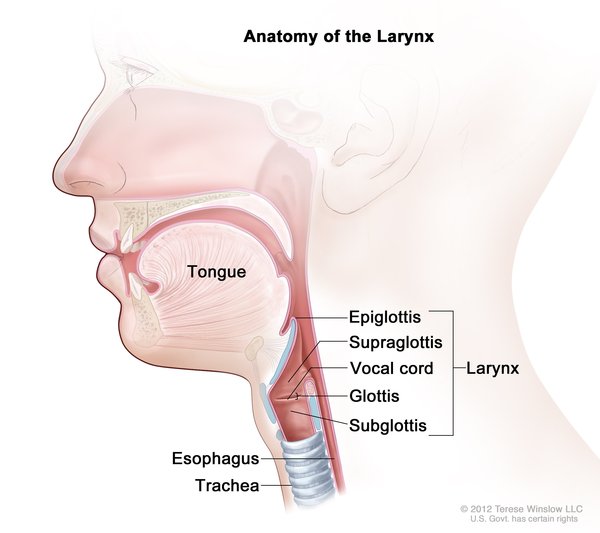
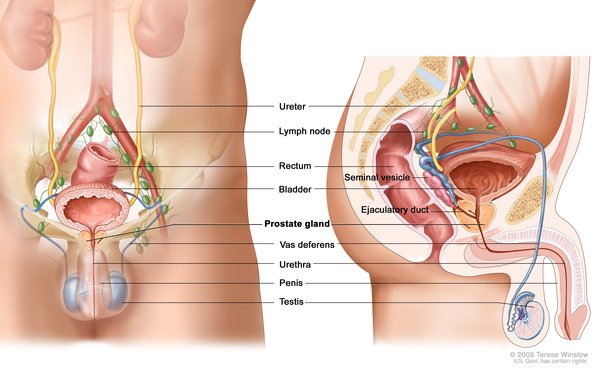
Nearly everyone with Opitz G/BBB syndrome has wide-spaced eyes (ocular hypertelorism). Affected individuals commonly have defects of the voice box (larynx), windpipe (trachea), or esophagus. These throat abnormalities can cause difficulty swallowing or breathing, in some cases resulting in recurrent pneumonia or life-threatening breathing problems. A common defect is a gap between the trachea and esophagus (laryngeal cleft) that allows food or fluids to enter the airway. The cleft can vary in size, and infants may struggle to breathe when feeding. Most males with Opitz G/BBB syndrome have genital abnormalities such as the urethra opening on the underside of the penis (hypospadias), undescended testes (cryptorchidism), an underdeveloped scrotum, or a scrotum divided into two lobes (bifid scrotum). These genital abnormalities can lead to problems in the urinary tract.

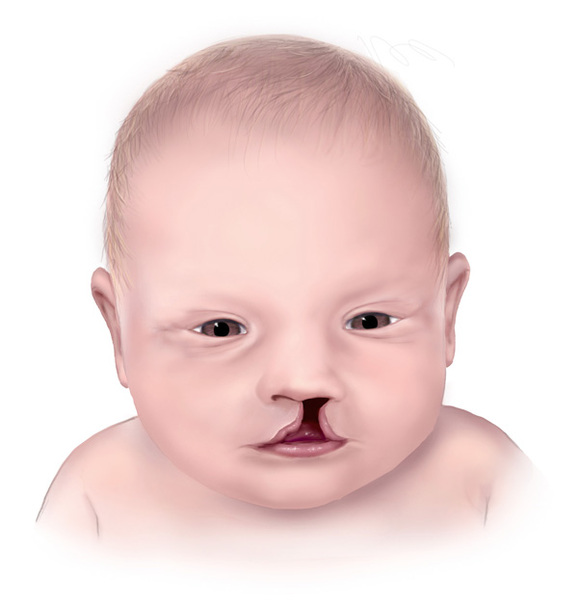
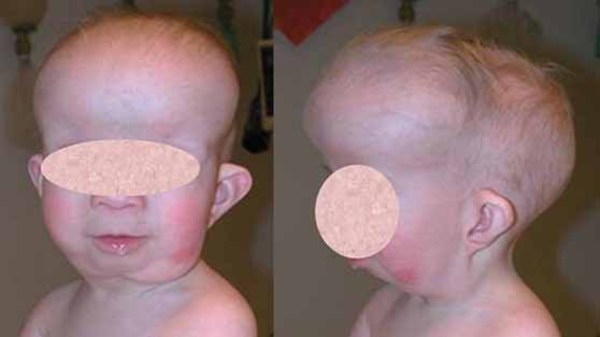
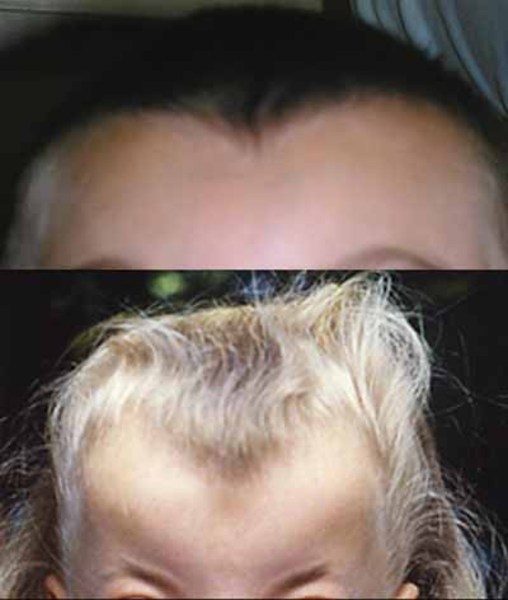
Mild intellectual disability and developmental delay occur in about 50 percent of people with Opitz G/BBB syndrome. Affected individuals have delayed motor skills, such as walking, speech delay, and learning difficulties. Some people with Opitz G/BBB syndrome have features of autistic spectrum disorders, which are characterized by impaired communication and socialization skills. About half of affected individuals also have an opening in the lip (cleft lip) with or without an opening in the roof of the mouth (cleft palate). Some have cleft palate without cleft lip. Less common features of Opitz G/BBB syndrome, affecting less than half of people with this disorder, include minor heart defects, an obstruction of the anal opening (imperforate anus), and brain defects such as a small or absent connection between the left and right halves of the brain (corpus callosum). Distinct facial features that may be seen in this disorder include a prominent forehead, widow's peak hairline, flat nasal bridge, thin upper lip, and low-set ears. These features vary among affected individuals, even within the same family.



Frequency
X-linked Opitz G/BBB syndrome is thought to affect 1 in 10,000 to 50,000 males, although it is likely that this condition is underdiagnosed.
The incidence of autosomal dominant Opitz G/BBB syndrome is unknown. It is part of a larger condition known as 22q11.2 deletion syndrome, which is estimated to affect 1 in 4,000 people.
Causes
X-linked Opitz G/BBB syndrome is caused by mutations in the MID1 gene. The MID1 gene provides instructions for making a protein called midline-1. This protein attaches (binds) to microtubules, which are rigid, hollow fibers that make up the cell's structural framework (the cytoskeleton). Microtubules help cells maintain their shape, assist in the process of cell division, and are essential for the movement of cells (cell migration). Midline-1 assists in recycling certain proteins that need to be reused instead of broken down. MID1 gene mutations lead to a decrease in midline-1 function, which prevents protein recycling. The resulting accumulation of proteins impairs microtubule function, leading to problems with cell division and migration. It is unclear how these changes disrupt normal development and cause the signs and symptoms of Opitz G/BBB syndrome.

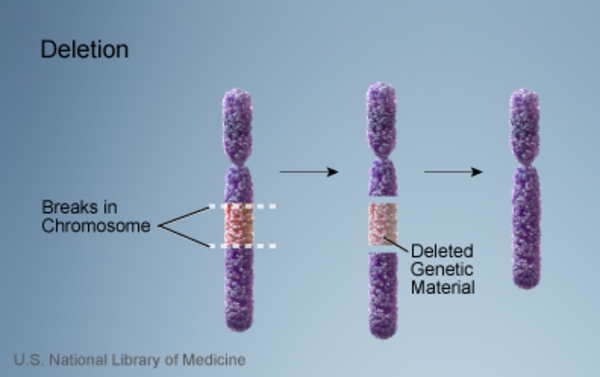
Autosomal dominant Opitz G/BBB syndrome is caused by changes in chromosome 22. Some affected individuals have a deletion of a small piece of chromosome 22, specifically at an area of the chromosome designated 22q11.2. Because this same region is deleted in another condition called 22q11.2 deletion syndrome, researchers often consider Opitz/GBBB syndrome caused by this genetic change to be a form of 22q11.2 deletion syndrome. It is not known which of the deleted genes contribute to the signs and symptoms of Opitz G/BBB syndrome.
In other people, autosomal dominant Opitz/GBBB syndrome is caused by a mutation in the SPECC1L gene, which is near the 22q11.2 region but is not in the area that is typically deleted in other individuals with autosomal dominant Opitz G/BBB syndrome or 22q11.2 deletion syndrome. The SPECC1L gene provides instructions for making a protein called cytospin-A. This protein interacts with components of the cytoskeleton and stabilizes microtubules, which is necessary for these fibers to regulate various cell processes including the movement of cells to their proper location (cell migration). Cytospin-A is particularly involved in the migration of cells that will form the facial features. Mutations in the SPECC1L gene result in the production of a protein with a decreased ability to interact with components of the cytoskeleton. As a result, microtubules are disorganized and cells have trouble migrating to their proper location. Because the SPECC1L gene plays a role in facial development, mutations in this gene likely account for the cleft lip and palate seen in some individuals with Opitz G/BBB syndrome, but it is unclear how SPECC1L gene mutations cause the other features of this disorder.
Some people with Opitz G/BBB syndrome do not have any of the genetic changes described above. The cause of the condition in these individuals is unknown.
Inheritance
When caused by mutations in the MID1 gene, Opitz G/BBB syndrome has an X-linked pattern of inheritance. It is considered X-linked because the MID1 gene is located on the X chromosome, one of the two sex chromosomes in each cell. In males, who have only one X chromosome, a mutation in the only copy of the gene in each cell is sufficient to cause the condition. In females, who have two copies of the X chromosome, one altered copy of the gene in each cell can lead to less severe features of the condition or may cause no symptoms at all. Because it is unlikely that females will have two altered copies of the MID1 gene, females with X-linked Opitz G/BBB syndrome typically have hypertelorism as the only sign of the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Rarely, Opitz G/BBB syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. These cases are caused by a mutation in the SPECC1L gene or by a deletion of genetic material from one copy of chromosome 22 in each cell. Males and females with autosomal dominant Opitz G/BBB syndrome usually have the same severity of symptoms.

In both types of Opitz G/BBB syndrome, some affected people inherit the genetic change from an affected parent. Other cases may result from new mutations. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- Hypertelorism with esophageal abnormalities and hypospadias
- Hypertelorism-hypospadias sydrome
- Hypospadias-dysphagia syndrome
- Opitz BBB syndrome
- Opitz BBB/G syndrome
- Opitz G syndrome
- Opitz syndrome
- Opitz-Frias syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- De Falco F, Cainarca S, Andolfi G, Ferrentino R, Berti C, Rodriguez Criado G, Rittinger O, Dennis N, Odent S, Rastogi A, Liebelt J, Chitayat D, Winter R, Jawanda H, Ballabio A, Franco B, Meroni G. X-linked Opitz syndrome: novel mutations in the MID1 gene and redefinition of the clinical spectrum. Am J Med Genet A. 2003 Jul 15;120A(2):222-8. doi: 10.1002/ajmg.a.10265. Citation on PubMed
- Fontanella B, Russolillo G, Meroni G. MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum Mutat. 2008 May;29(5):584-94. doi: 10.1002/humu.20706. Citation on PubMed
- Fryburg JS, Lin KY, Golden WL. Chromosome 22q11.2 deletion in a boy with Opitz (G/BBB) syndrome. Am J Med Genet. 1996 Mar 29;62(3):274-5. doi: 10.1002/(SICI)1096-8628(19960329)62:33.0.CO;2-H. Citation on PubMed
- Han X, Du H, Massiah MA. Detection and characterization of the in vitro e3 ligase activity of the human MID1 protein. J Mol Biol. 2011 Apr 8;407(4):505-20. doi: 10.1016/j.jmb.2011.01.048. Epub 2011 Feb 4. Citation on PubMed
- Kruszka P, Li D, Harr MH, Wilson NR, Swarr D, McCormick EM, Chiavacci RM, Li M, Martinez AF, Hart RA, McDonald-McGinn DM, Deardorff MA, Falk MJ, Allanson JE, Hudson C, Johnson JP, Saadi I, Hakonarson H, Muenke M, Zackai EH. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J Med Genet. 2015 Feb;52(2):104-10. doi: 10.1136/jmedgenet-2014-102677. Epub 2014 Nov 20. Citation on PubMed or Free article on PubMed Central
- McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J. Autosomal dominant "Opitz" GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet. 1995 Oct 23;59(1):103-13. doi: 10.1002/ajmg.1320590122. Citation on PubMed
- Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 1997 Nov;17(3):285-91. doi: 10.1038/ng1197-285. Citation on PubMed
- Robin NH, Opitz JM, Muenke M. Opitz G/BBB syndrome: clinical comparisons of families linked to Xp22 and 22q, and a review of the literature. Am J Med Genet. 1996 Mar 29;62(3):305-17. doi: 10.1002/(SICI)1096-8628(19960329)62:33.0.CO;2-N. No abstract available. Citation on PubMed
- Schweiger S, Schneider R. The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. Bioessays. 2003 Apr;25(4):356-66. doi: 10.1002/bies.10256. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.