

Description
Oculodentodigital dysplasia is a condition that affects many parts of the body, particularly the eyes (oculo-), teeth (dento-), and fingers (digital). Common features in people with this condition are small eyes (microphthalmia) and other eye abnormalities that can lead to vision loss. Affected individuals also frequently have tooth abnormalities, such as small or missing teeth, weak enamel, multiple cavities, and early tooth loss. Other common features of this condition include a thin nose and webbing of the skin (syndactyly) between the fourth and fifth fingers.

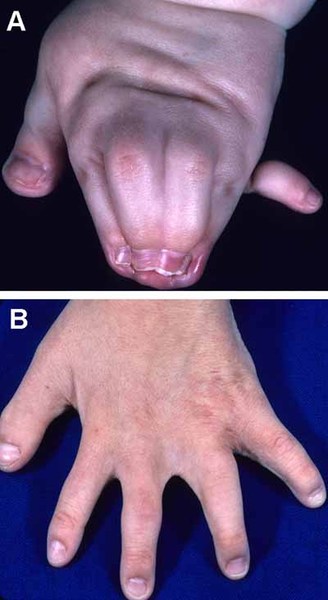
Less common features of oculodentodigital dysplasia include sparse hair growth (hypotrichosis), brittle nails, an unusual curvature of the fingers (camptodactyly), syndactyly of the toes, small head size (microcephaly), and an opening in the roof of the mouth (cleft palate). Some affected individuals experience neurological problems such as a lack of bladder or bowel control, difficulty coordinating movements (ataxia), abnormal muscle stiffness (spasticity), hearing loss, and impaired speech (dysarthria). A few people with oculodentodigital dysplasia also have a skin condition called palmoplantar keratoderma. Palmoplantar keratoderma causes the skin on the palms and the soles of the feet to become thick, scaly, and calloused.

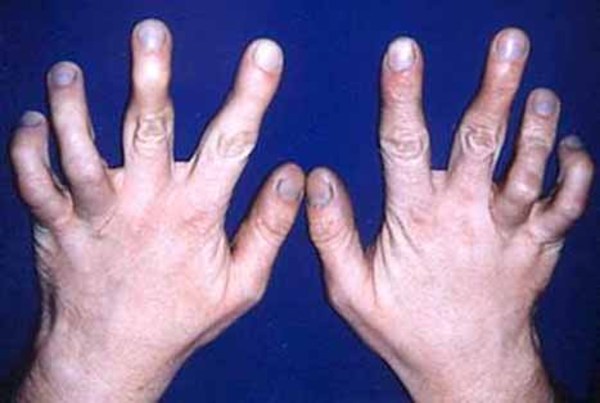
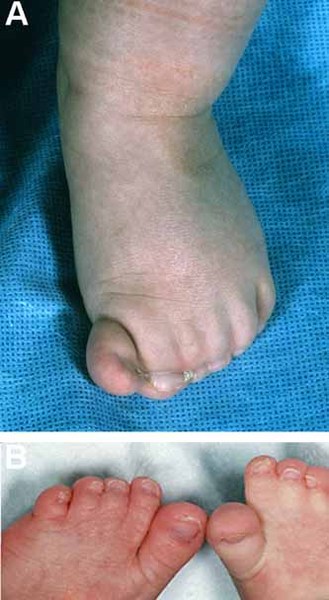


Some features of oculodentodigital dysplasia are evident at birth, while others become apparent with age.
Frequency
The exact incidence of oculodentodigital dysplasia is unknown. It has been diagnosed in fewer than 1,000 people worldwide. More cases are likely undiagnosed.
Causes
Mutations in the GJA1 gene cause oculodentodigital dysplasia. The GJA1 gene provides instructions for making a protein called connexin 43. This protein forms one part (a subunit) of channels called gap junctions, which allow direct communication between cells. Gap junctions formed by connexin 43 proteins are found in many tissues throughout the body.
GJA1 gene mutations result in abnormal connexin 43 proteins. Channels formed with abnormal proteins are often permanently closed. Some mutations prevent connexin 43 proteins from traveling to the cell surface where they are needed to form channels between cells. Impaired functioning of these channels disrupts cell-to-cell communication, which likely interferes with normal cell growth and cell specialization, processes that determine the shape and function of many different parts of the body. These developmental problems cause the signs and symptoms of oculodentodigital dysplasia.
Inheritance
Most cases of oculodentodigital dysplasia are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Less commonly, oculodentodigital dysplasia can be inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Fewer than ten cases of autosomal recessive oculodentodigital dysplasia have been reported.

Other Names for This Condition
- Oculo-dento-digital dysplasia
- Oculo-dento-osseous dysplasia
- Oculodentodigital syndrome
- Oculodentoosseous dysplasia
- ODD syndrome
- ODDD
- ODOD
- Osseous-oculo-dental dysplasia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Debeer P, Van Esch H, Huysmans C, Pijkels E, De Smet L, Van de Ven W, Devriendt K, Fryns JP. Novel GJA1 mutations in patients with oculo-dento-digital dysplasia (ODDD). Eur J Med Genet. 2005 Oct-Dec;48(4):377-87. doi: 10.1016/j.ejmg.2005.05.003. Citation on PubMed
- Frasson M, Calixto N, Cronemberger S, de Aguiar RA, Leao LL, de Aguiar MJ. Oculodentodigital dysplasia: study of ophthalmological and clinical manifestations in three boys with probably autosomal recessive inheritance. Ophthalmic Genet. 2004 Sep;25(3):227-36. doi: 10.1080/13816810490513424. Citation on PubMed
- Joss SK, Ghazawy S, Tomkins S, Ahmed M, Bradbury J, Sheridan E. Variable expression of neurological phenotype in autosomal recessive oculodentodigital dysplasia of two sibs and review of the literature. Eur J Pediatr. 2008 Mar;167(3):341-5. doi: 10.1007/s00431-007-0468-1. Epub 2007 May 3. Citation on PubMed
- Laird DW. Closing the gap on autosomal dominant connexin-26 and connexin-43 mutants linked to human disease. J Biol Chem. 2008 Feb 8;283(6):2997-3001. doi: 10.1074/jbc.R700041200. Epub 2007 Dec 18. Citation on PubMed
- Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006 Mar 15;394(Pt 3):527-43. doi: 10.1042/BJ20051922. Citation on PubMed or Free article on PubMed Central
- Loddenkemper T, Grote K, Evers S, Oelerich M, Stogbauer F. Neurological manifestations of the oculodentodigital dysplasia syndrome. J Neurol. 2002 May;249(5):584-95. doi: 10.1007/s004150200068. Citation on PubMed
- Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, Jabs EW. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003 Feb;72(2):408-18. doi: 10.1086/346090. Epub 2002 Nov 27. Citation on PubMed or Free article on PubMed Central
- Shibayama J, Paznekas W, Seki A, Taffet S, Jabs EW, Delmar M, Musa H. Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ Res. 2005 May 27;96(10):e83-91. doi: 10.1161/01.RES.0000168369.79972.d2. Epub 2005 May 5. Citation on PubMed
- van Steensel MA, Spruijt L, van der Burgt I, Bladergroen RS, Vermeer M, Steijlen PM, van Geel M. A 2-bp deletion in the GJA1 gene is associated with oculo-dento-digital dysplasia with palmoplantar keratoderma. Am J Med Genet A. 2005 Jan 15;132A(2):171-4. doi: 10.1002/ajmg.a.30412. Citation on PubMed
- Vreeburg M, de Zwart-Storm EA, Schouten MI, Nellen RG, Marcus-Soekarman D, Devies M, van Geel M, van Steensel MA. Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. Am J Med Genet A. 2007 Feb 15;143(4):360-3. doi: 10.1002/ajmg.a.31558. Citation on PubMed
- Wiest T, Herrmann O, Stogbauer F, Grasshoff U, Enders H, Koch MJ, Grond-Ginsbach C, Schwaninger M. Clinical and genetic variability of oculodentodigital dysplasia. Clin Genet. 2006 Jul;70(1):71-2. doi: 10.1111/j.1399-0004.2006.00631.x. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.