

Description
Noonan syndrome with multiple lentigines (formerly called LEOPARD syndrome) is a condition that affects many areas of the body. As the condition name suggests, Noonan syndrome with multiple lentigines is very similar to a condition called Noonan syndrome, and it can be difficult to tell the two disorders apart in early childhood. However, the features of these two conditions differ later in life. The characteristic features of Noonan syndrome with multiple lentigines include brown skin spots called lentigines that are similar to freckles, heart defects, widely spaced eyes (ocular hypertelorism), a sunken chest (pectus excavatum) or protruding chest (pectus carinatum), and short stature. These features vary, however, even among affected individuals in the same family. Not all individuals with Noonan syndrome with multiple lentigines have all the characteristic features of this condition.

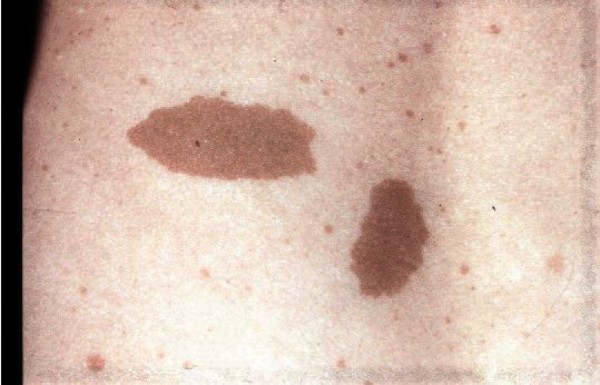
The lentigines seen in Noonan syndrome with multiple lentigines typically first appear in mid-childhood, mostly on the face, neck, and upper body. Affected individuals may have thousands of small dark brown skin spots by the time they reach puberty. Unlike freckles, the appearance of lentigines has nothing to do with sun exposure. In addition to lentigines, people with this condition may have lighter brown skin spots called café-au-lait spots. Café-au-lait spots tend to develop before the lentigines, appearing within the first year of life in most affected people.
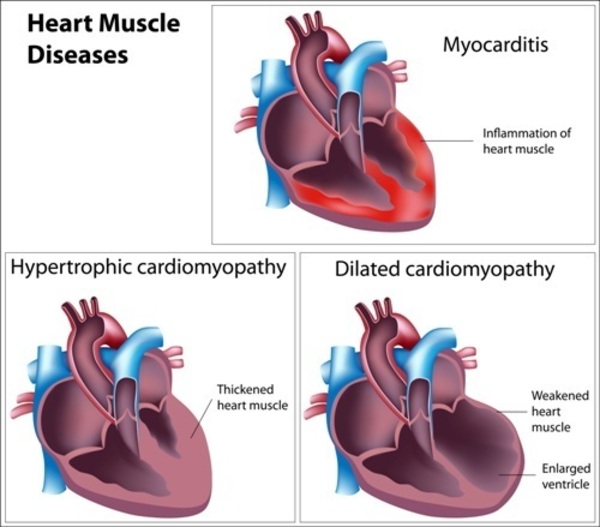
Of the people with Noonan syndrome with multiple lentigines who have heart defects, about 80 percent have hypertrophic cardiomyopathy, which is a thickening of the heart muscle that forces the heart to work harder to pump blood. The hypertrophic cardiomyopathy most often affects the lower left chamber of the heart (the left ventricle). Up to 20 percent of people with Noonan syndrome with multiple lentigines who have heart problems have a narrowing of the artery from the heart to the lungs (pulmonary stenosis).
People with Noonan syndrome with multiple lentigines can have a distinctive facial appearance. In addition to ocular hypertelorism, affected individuals may have droopy eyelids (ptosis), thick lips, and low-set ears. Affected individuals also usually have an abnormal appearance of the chest; they either have pectus excavatum or pectus carinatum.
At birth, people with Noonan syndrome with multiple lentigines are typically of normal weight and height, but in some, growth slows over time. This slow growth results in affected individuals being shorter than average, although less than half of people with Noonan syndrome with multiple lentigines have significantly short stature.
Other signs and symptoms of Noonan syndrome with multiple lentigines include hearing loss caused by abnormalities in the inner ear (sensorineural deafness), mild intellectual disability, and extra folds of skin on the back of the neck. Affected males often have genital abnormalities, which can include undescended testes (cryptorchidism) and a urethra that opens on the underside of the penis (hypospadias). These abnormalities may reduce the ability to have biological children (decreased fertility). Females with Noonan syndrome with multiple lentigines may have poorly developed ovaries and delayed puberty.

Noonan syndrome with multiple lentigines is one of a group of related conditions collectively known as RASopathies. These conditions all have similar signs and symptoms and are caused by changes in the same cell signaling pathway. In addition to Noonan syndrome with multiple lentigines, the RASopathies include Noonan syndrome, cardiofaciocutaneous syndrome, Costello syndrome, neurofibromatosis type 1, and Legius syndrome.
Frequency
Noonan syndrome with multiple lentigines is thought to be a rare condition; approximately 200 cases have been reported worldwide.
Causes
Variants (also known as mutations) in one of several genes can cause Noonan syndrome with multiple lentigines. Approximately 85 percent of individuals with this condition have mutations in the PTPN11 gene. Another 10 percent have mutations in the RAF1 gene. In rare cases, mutations in the BRAF or MAP2K1 gene have been found to cause this condition. The remaining individuals with Noonan syndrome with multiple lentigines do not have an identified mutation in any of these four genes. In these individuals, the cause of the condition is unknown.
The PTPN11, RAF1, BRAF, and MAP2K1 genes all provide instructions for making proteins that are involved in important signaling pathways needed for the proper formation of several types of tissue during development. These proteins also play roles in the regulation of cell division , cell movement (migration), and cell differentiation (the process by which cells mature to carry out specific functions).
, cell movement (migration), and cell differentiation (the process by which cells mature to carry out specific functions).
A mutation in the PTPN11, RAF1, BRAF, or MAP2K1 gene leads to the production of a protein that functions abnormally, which impairs the protein's ability to respond to cell signals. A disruption in the regulation of systems that control cell growth and division leads to the characteristic features of Noonan syndrome with multiple lentigines.
Inheritance
Noonan syndrome with multiple lentigines
is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the condition.
Some affected people inherit the variation from one affected parent . Other cases result from new variants in the gene
. Other cases result from new variants in the gene and occur in people with no history of the condition in their family.
and occur in people with no history of the condition in their family.
Other Names for This Condition
- Cardio-cutaneous syndrome
- Cardiomyopathic lentiginosis
- Diffuse lentiginosis
- Lentiginosis profusa
- LEOPARD syndrome
- Moynahan syndrome
- Multiple lentigines syndrome
- NSML
- Progressive cardiomyopathic lentiginosis
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Kato H, Yoshida R, Tsukamoto K, Suga H, Eto H, Higashino T, Araki J, Ogata T, Yoshimura K. Familial cases of atypical clinical features genetically diagnosed as LEOPARD syndrome (multiple lentigines syndrome). Int J Dermatol. 2010 Oct;49(10):1146-51. doi: 10.1111/j.1365-4632.2010.04559.x. Citation on PubMed
- Limongelli G, Pacileo G, Marino B, Digilio MC, Sarkozy A, Elliott P, Versacci P, Calabro P, De Zorzi A, Di Salvo G, Syrris P, Patton M, McKenna WJ, Dallapiccola B, Calabro R. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 2007 Aug 15;100(4):736-41. doi: 10.1016/j.amjcard.2007.03.093. Epub 2007 Jun 27. Citation on PubMed
- Nishi E, Mizuno S, Nanjo Y, Niihori T, Fukushima Y, Matsubara Y, Aoki Y, Kosho T. A novel heterozygous MAP2K1 mutation in a patient with Noonan syndrome with multiple lentigines. Am J Med Genet A. 2015 Feb;167A(2):407-11. doi: 10.1002/ajmg.a.36842. Epub 2014 Nov 25. Citation on PubMed
- Santoro C, Pacileo G, Limongelli G, Scianguetta S, Giugliano T, Piluso G, Ragione FD, Cirillo M, Mirone G, Perrotta S. LEOPARD syndrome: clinical dilemmas in differential diagnosis of RASopathies. BMC Med Genet. 2014 Apr 26;15:44. doi: 10.1186/1471-2350-15-44. Citation on PubMed or Free article on PubMed Central
- Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008 May 27;3:13. doi: 10.1186/1750-1172-3-13. Citation on PubMed or Free article on PubMed Central
- Stevenson DA, Schill L, Schoyer L, Andresen BS, Bakker A, Bayrak-Toydemir P, Burkitt-Wright E, Chatfield K, Elefteriou F, Elgersma Y, Fisher MJ, Franz D, Gelb BD, Goriely A, Gripp KW, Hardan AY, Keppler-Noreuil KM, Kerr B, Korf B, Leoni C, McCormick F, Plotkin SR, Rauen KA, Reilly K, Roberts A, Sandler A, Siegel D, Walsh K, Widemann BC. The Fourth International Symposium on Genetic Disorders of the Ras/MAPK pathway. Am J Med Genet A. 2016 Aug;170(8):1959-66. doi: 10.1002/ajmg.a.37723. Epub 2016 May 7. Citation on PubMed
- Yu ZH, Zhang RY, Walls CD, Chen L, Zhang S, Wu L, Liu S, Zhang ZY. Molecular basis of gain-of-function LEOPARD syndrome-associated SHP2 mutations. Biochemistry. 2014 Jul 1;53(25):4136-51. doi: 10.1021/bi5002695. Epub 2014 Jun 17. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.