

Description
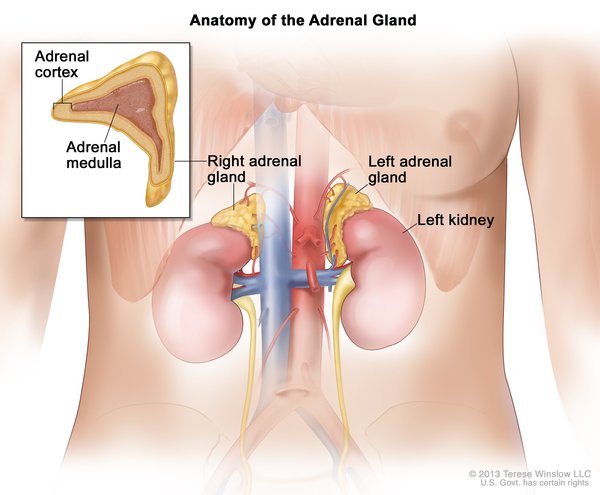
Paraganglioma is a type of noncancerous (benign) tumor that occurs in structures called paraganglia. Paraganglia are groups of cells that are found near nerve cell bunches called ganglia. Paragangliomas are usually found in the head, neck, or torso. However, a type of paraganglioma known as pheochromocytoma develops in the adrenal glands. Adrenal glands are located on top of each kidney and produce hormones in response to stress. Most people with paraganglioma develop only one tumor in their lifetime.
Some people develop a paraganglioma or pheochromocytoma as part of a hereditary syndrome that may affect other organs and tissues in the body. However, the tumors often are not associated with any syndromes, in which case the condition is called nonsyndromic paraganglioma or pheochromocytoma.
Pheochromocytomas and some other paragangliomas are associated with ganglia of the sympathetic nervous system. The sympathetic nervous system controls the "fight-or-flight" response, a series of changes in the body due to hormones released in response to stress. Although most sympathetic paragangliomas are pheochromocytomas, some are found outside the adrenal glands, usually in the abdomen, and are called extra-adrenal paragangliomas. Most sympathetic paragangliomas, including pheochromocytomas, produce hormones called catecholamines, such as epinephrine (adrenaline) or norepinephrine. These excess catecholamines can cause signs and symptoms such as high blood pressure (hypertension), episodes of rapid heartbeat (palpitations), headaches, or sweating.
Most paragangliomas are associated with ganglia of the parasympathetic nervous system, which controls involuntary body functions such as digestion and saliva formation. Parasympathetic paragangliomas, typically found in the head and neck, usually do not produce hormones. However, large tumors may cause signs and symptoms such as coughing, hearing loss in one ear, or difficulty swallowing.
Although most paragangliomas and pheochromocytomas are noncancerous, some can become cancerous (malignant) and spread to other parts of the body (metastasize). Extra-adrenal paragangliomas become malignant more often than other types of paraganglioma or pheochromocytoma.
Frequency
It is estimated that the prevalence of pheochromocytoma is 1 in 500,000 people, and the prevalence of other paragangliomas is 1 in 1 million people. These statistics include syndromic and nonsyndromic paraganglioma and pheochromocytoma.
Causes
The VHL, RET, SDHB, and SDHD genes can be mutated in both syndromic and nonsyndromic forms of paraganglioma and pheochromocytoma. Mutations in at least three additional genes, TMEM127, SDHA, and KIF1B, have been identified in people with the nonsyndromic form of these conditions. Gene mutations increase the risk of developing paraganglioma or pheochromocytoma by affecting control of cell growth and division.
Mutations in the VHL, SDHA, SDHB, and SDHD genes increase the risk of developing nonsyndromic paraganglioma or pheochromocytoma. The protein produced from the VHL gene helps break down other, unneeded proteins, including a protein called HIF that stimulates cell division and blood vessel formation under certain cellular conditions. The proteins produced from the SDHA, SDHB, and SDHD genes are each pieces (subunits) of an enzyme that is important for energy production in the cell. This enzyme also plays a role in the breakdown of the HIF protein. Mutations in the VHL, SDHA, SDHB, and SDHD genes stabilize the HIF protein, causing it to build up in cells. Excess HIF protein stimulates cells to divide and triggers the production of blood vessels when they are not needed. Rapid and uncontrolled cell division, along with the formation of new blood vessels, can lead to the development of tumors.
Mutations in the RET gene have been found in nonsyndromic pheochromocytoma in addition to a pheochromocytoma-predisposing syndrome. The protein produced from the RET gene is involved in signaling within cells that can stimulate cell division or maturation. Mutations in the RET gene overactivate the protein's signaling function, which can trigger cell growth and division in the absence of signals from outside the cell. This unchecked cell division can lead to the formation of tumors in the adrenal glands.
Mutations in the TMEM127 gene have been identified most commonly in people with nonsyndromic pheochromocytoma and are rarely seen in people with other paraganglioma. The TMEM127 protein normally controls a signaling pathway that induces cell growth and survival. Studies suggest that mutations in the TMEM127 gene lead to abnormal activation of cell growth, which may cause tumor formation.
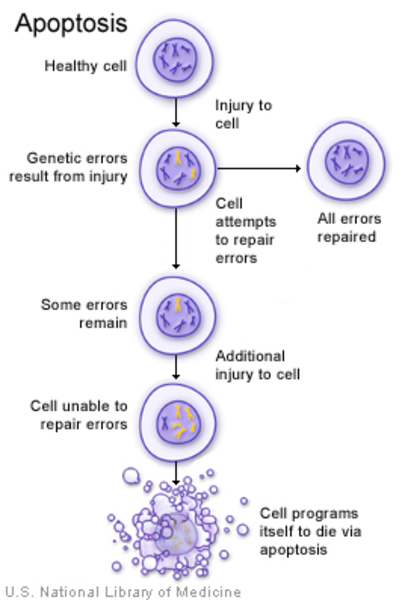
Mutations in the KIF1B gene have been reported in nonsyndromic pheochromocytoma. Studies suggest that these mutations impair the function of the KIF1B protein, which normally triggers cells to self-destruct in a process called apoptosis. When apoptosis is impaired, cells grow and divide too quickly or in an uncontrolled way, potentially leading to tumor formation.
Many people with nonsyndromic paraganglioma or pheochromocytoma do not have a mutation in any of the genes associated with the conditions. It is likely that other, unidentified genes also predispose to development of paraganglioma or pheochromocytoma.
Inheritance
Nonsyndromic paraganglioma can be inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to increase the risk of developing a paraganglioma or pheochromocytoma. People with mutations in the gene inherit an increased risk of this condition, not the condition itself. Not all people with this condition have a mutation in the gene, and not all people with a gene mutation will develop the disorder.

Most cases of nonsyndromic paraganglioma and pheochromocytoma are considered sporadic, which means the tumors occur in people with no history of the disorder in their family.
Other Names for This Condition
- Chemodectoma
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, Jouanno E, Jeunemaitre X, Benit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-Roqueplo AP. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010 Aug 1;19(15):3011-20. doi: 10.1093/hmg/ddq206. Epub 2010 May 18. Citation on PubMed or Free article on PubMed Central
- Burnichon N, Vescovo L, Amar L, Libe R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J, Plouin PF, Jeunemaitre X, Favier J, Gimenez-Roqueplo AP. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011 Oct 15;20(20):3974-85. doi: 10.1093/hmg/ddr324. Epub 2011 Jul 22. Citation on PubMed
- Crona J, Taieb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr Rev. 2017 Dec 1;38(6):489-515. doi: 10.1210/er.2017-00062. Citation on PubMed or Free article on PubMed Central
- Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, Badoual C, Gadessaud N, Venisse A, Bayley JP, van Dooren MF, de Herder WW, Tissier F, Plouin PF, van Nederveen FH, Dinjens WN, Gimenez-Roqueplo AP, de Krijger RR. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011 Sep;96(9):E1472-6. doi: 10.1210/jc.2011-1043. Epub 2011 Jul 13. Citation on PubMed
- Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002 May 9;346(19):1459-66. doi: 10.1056/NEJMoa020152. Citation on PubMed
- Neumann HP, Sullivan M, Winter A, Malinoc A, Hoffmann MM, Boedeker CC, Bertz H, Walz MK, Moeller LC, Schmid KW, Eng C. Germline mutations of the TMEM127 gene in patients with paraganglioma of head and neck and extraadrenal abdominal sites. J Clin Endocrinol Metab. 2011 Aug;96(8):E1279-82. doi: 10.1210/jc.2011-0114. Epub 2011 May 25. Citation on PubMed
- Opocher G, Schiavi F. Genetics of pheochromocytomas and paragangliomas. Best Pract Res Clin Endocrinol Metab. 2010 Dec;24(6):943-56. doi: 10.1016/j.beem.2010.05.001. Citation on PubMed
- Qin Y, Buddavarapu K, Dahia PL. Pheochromocytomas: from genetic diversity to new paradigms. Horm Metab Res. 2009 Sep;41(9):664-71. doi: 10.1055/s-0029-1215590. Epub 2009 Apr 23. Citation on PubMed
- Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, Boaretto F, Opocher G, Toledo RA, Toledo SP, Stiles C, Aguiar RC, Dahia PL. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010 Mar;42(3):229-33. doi: 10.1038/ng.533. Epub 2010 Feb 14. Citation on PubMed or Free article on PubMed Central
- Yao L, Schiavi F, Cascon A, Qin Y, Inglada-Perez L, King EE, Toledo RA, Ercolino T, Rapizzi E, Ricketts CJ, Mori L, Giacche M, Mendola A, Taschin E, Boaretto F, Loli P, Iacobone M, Rossi GP, Biondi B, Lima-Junior JV, Kater CE, Bex M, Vikkula M, Grossman AB, Gruber SB, Barontini M, Persu A, Castellano M, Toledo SP, Maher ER, Mannelli M, Opocher G, Robledo M, Dahia PL. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA. 2010 Dec 15;304(23):2611-9. doi: 10.1001/jama.2010.1830. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.