

Description
Neuromyelitis optica is an autoimmune disorder that affects the nerves of the eyes and the central nervous system, which includes the brain and spinal cord. Autoimmune disorders occur when the immune system malfunctions and attacks the body's own tissues and organs. In neuromyelitis optica, the autoimmune attack causes inflammation of the nerves, and the resulting damage leads to the signs and symptoms of the condition.
Neuromyelitis optica is characterized by optic neuritis, which is inflammation of the nerve that carries information from the eye to the brain (optic nerve). Optic neuritis causes eye pain and vision loss, which can occur in one or both eyes.
Neuromyelitis optica is also characterized by transverse myelitis, which is inflammation of the spinal cord. The inflammation associated with transverse myelitis damages the spinal cord, causing a lesion that often extends the length of three or more bones of the spine (vertebrae). In addition, myelin, which is the covering that protects nerves and promotes the efficient transmission of nerve impulses, can be damaged. Transverse myelitis causes weakness, numbness, and paralysis of the arms and legs. Other effects of spinal cord damage can include disturbances in sensations, loss of bladder and bowel control, uncontrollable hiccupping, and nausea. In addition, muscle weakness may make breathing difficult and can cause life-threatening respiratory failure in people with neuromyelitis optica.

There are two forms of neuromyelitis optica, the relapsing form and the monophasic form. The relapsing form is most common. This form is characterized by recurrent episodes of optic neuritis and transverse myelitis. These episodes can be months or years apart, and there is usually partial recovery between episodes. However, most affected individuals eventually develop permanent muscle weakness and vision impairment that persist even between episodes. For unknown reasons, approximately nine times more women than men have the relapsing form. The monophasic form, which is less common, causes a single episode of neuromyelitis optica that can last several months. People with this form of the condition can also have lasting muscle weakness or paralysis and vision loss. This form affects men and women equally. The onset of either form of neuromyelitis optica can occur anytime from childhood to adulthood, although the condition most frequently begins in a person's forties.
Approximately one-quarter of individuals with neuromyelitis optica have signs or symptoms of another autoimmune disorder such as myasthenia gravis, systemic lupus erythematosus, or Sjögren syndrome. Some scientists believe that a condition described in Japanese patients as optic-spinal multiple sclerosis (or opticospinal multiple sclerosis) that affects the nerves of the eyes and central nervous system is the same as neuromyelitis optica.
Frequency
Neuromyelitis optica affects approximately 1 to 2 per 100,000 people worldwide. Women are affected by this condition more frequently than men.
Causes
No genes associated with neuromyelitis optica have been identified. However, a small percentage of people with this condition have a family member who is also affected, which indicates that there may be one or more genetic changes that increase susceptibility. It is thought that the inheritance of this condition is complex and that many environmental and genetic factors are involved in the development of the condition.
The aquaporin-4 protein (AQP4), a normal protein in the body, plays a role in neuromyelitis optica. The aquaporin-4 protein is found in several body systems but is most abundant in tissues of the central nervous system. Approximately 70 percent of people with this disorder produce an immune protein called an antibody that attaches (binds) to the aquaporin-4 protein. Antibodies normally bind to specific foreign particles and germs, marking them for destruction, but the antibody in people with neuromyelitis optica attacks a normal human protein; this type of antibody is called an autoantibody. The autoantibody in this condition is called NMO-IgG or anti-AQP4.
The binding of the NMO-IgG autoantibody to the aquaporin-4 protein turns on (activates) the complement system, which is a group of immune system proteins that work together to destroy pathogens, trigger inflammation, and remove debris from cells and tissues. Complement activation leads to the inflammation of the optic nerve and spinal cord that is characteristic of neuromyelitis optica, resulting in the signs and symptoms of the condition.
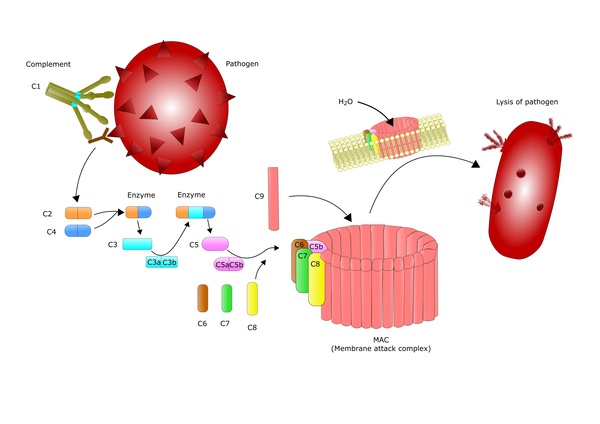
The levels of the NMO-IgG autoantibody are high during episodes of neuromyelitis optica, and the levels decrease between episodes with treatment of the disorder. However, it is unclear what triggers episodes to begin or end.
Inheritance
Neuromyelitis optica is usually not inherited. Rarely, this condition is passed through generations in families, but the inheritance pattern is unknown.
Other Names for This Condition
- Devic disease
- Devic neuromyelitis optica
- Devic syndrome
- Devic's disease
- Optic-spinal MS
- Opticospinal MS
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007 Dec 11;69(24):2221-31. doi: 10.1212/01.WNL.0000289761.64862.ce. Epub 2007 Oct 10. Citation on PubMed
- Mata S, Lolli F. Neuromyelitis optica: an update. J Neurol Sci. 2011 Apr 15;303(1-2):13-21. doi: 10.1016/j.jns.2011.01.002. Citation on PubMed
- Matiello M, Kim HJ, Kim W, Brum DG, Barreira AA, Kingsbury DJ, Plant GT, Adoni T, Weinshenker BG. Familial neuromyelitis optica. Neurology. 2010 Jul 27;75(4):310-5. doi: 10.1212/WNL.0b013e3181ea9f15. Citation on PubMed or Free article on PubMed Central
- Matiello M, Schaefer-Klein JL, Hebrink DD, Kingsbury DJ, Atkinson EJ, Weinshenker BG; NMO Genetics Collaborators. Genetic analysis of aquaporin-4 in neuromyelitis optica. Neurology. 2011 Sep 20;77(12):1149-55. doi: 10.1212/WNL.0b013e31822f045b. Epub 2011 Sep 7. Citation on PubMed
- National Institute of Neurological Disorders and Stroke: Neuromyelitis Optica Information Page
- Sato DK, Lana-Peixoto MA, Fujihara K, de Seze J. Clinical spectrum and treatment of neuromyelitis optica spectrum disorders: evolution and current status. Brain Pathol. 2013 Nov;23(6):647-60. doi: 10.1111/bpa.12087. Citation on PubMed
- Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, Watanabe S, Shiga Y, Kanaoka C, Fujimori J, Sato S, Itoyama Y. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007 May;130(Pt 5):1235-43. doi: 10.1093/brain/awm062. Epub 2007 Apr 19. Citation on PubMed
- Veszeli N, Fust G, Csuka D, Trauninger A, Bors L, Rozsa C, Nagy Z, Jobbagy Z, Eizler K, Prohaszka Z, Varga L, Illes Z. A systematic analysis of the complement pathways in patients with neuromyelitis optica indicates alteration but no activation during remission. Mol Immunol. 2014 Feb;57(2):200-9. doi: 10.1016/j.molimm.2013.09.010. Epub 2013 Oct 26. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.