

Description
Nemaline myopathy is a disorder that primarily affects skeletal muscles, which are muscles that the body uses for movement. People with nemaline myopathy have muscle weakness (myopathy) throughout the body, but it is typically most severe in the muscles of the face; neck; trunk; and other muscles close to the center of the body (proximal muscles), such as those of the upper arms and legs. This weakness can worsen over time. Affected individuals may have feeding and swallowing difficulties, foot deformities, abnormal curvature of the spine (scoliosis), and joint deformities (contractures). Most people with nemaline myopathy are able to walk, although some affected children may begin walking later than usual. As the condition progresses, some people may require wheelchair assistance. In severe cases, the muscles used for breathing are affected and life-threatening breathing difficulties can occur.

Nemaline myopathy is divided into six types. In order of decreasing severity, the types are: severe congenital, Amish, intermediate congenital, typical congenital, childhood-onset, and adult-onset. The types are distinguished by the age when symptoms first appear and the severity of symptoms; however, there is overlap among the various types. The severe congenital type is the most life-threatening. Most individuals with this type do not survive past early childhood due to respiratory failure. The Amish type solely affects the Old Order Amish population of Pennsylvania and is typically fatal in early childhood. The most common type of nemaline myopathy is the typical congenital type, which is characterized by muscle weakness and feeding problems beginning in infancy. Most of these individuals do not have severe breathing problems and can walk unassisted. People with the childhood-onset type usually develop muscle weakness in adolescence. The adult-onset type is the mildest of all the various types. People with this type usually develop muscle weakness between ages 20 and 50.
Frequency
Nemaline myopathy has an estimated incidence of 1 in 50,000 individuals.
Causes
Mutations in one of many genes can cause nemaline myopathy. These genes provide instructions for producing proteins that play important roles in skeletal muscles. Within skeletal muscle cells, these proteins are found in structures called sarcomeres. Sarcomeres are necessary for muscles to tense (contract). Many of the proteins associated with nemaline myopathy interact within the sarcomere to facilitate muscle contraction. When the skeletal muscle cells of people with nemaline myopathy are stained and viewed under a microscope, these cells usually appear abnormal. These abnormal muscle cells contain rod-like structures called nemaline bodies.
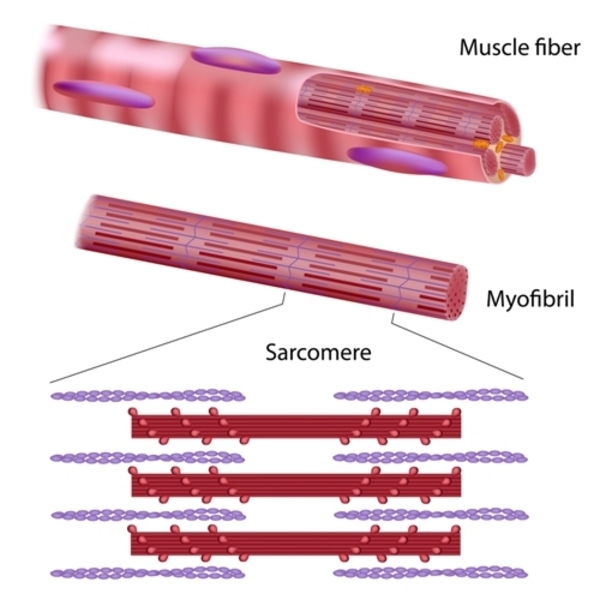
Most cases of nemaline myopathy with a known genetic cause result from mutations in one of two genes, NEB or ACTA1. NEB gene mutations account for about 50 percent of all cases of nemaline myopathy and ACTA1 gene mutations account for 15 to 25 percent of all cases. When nemaline myopathy is caused by NEB gene mutations, signs and symptoms are typically present at birth or beginning in early childhood. When nemaline myopathy is caused by ACTA1 gene mutations, the condition's severity and age of onset vary widely. Mutations in the other genes associated with nemaline myopathy each account for only a small percentage of cases.
Mutations in any of the genes associated with nemaline myopathy lead to disorganization of the proteins found in the sarcomeres of skeletal muscles. The disorganized proteins cannot interact normally, which disrupts muscle contraction. Inefficient muscle contraction leads to muscle weakness and the other features of nemaline myopathy.
Some individuals with nemaline myopathy do not have an identified mutation. The genetic cause of the disorder is unknown in these individuals.
Inheritance
Nemaline myopathy is usually inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Less often, this condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new mutations in the gene and occur in people with no history of the disorder in their family.

Other Names for This Condition
- Myopathies, nemaline
- Myopathy, nemaline
- Nemaline body disease
- Nemaline rod disease
- Rod body disease
- Rod myopathy
- Rod-body myopathy
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Actin accumulation myopathy

- Genetic Testing Registry: Congenital myopathy 23
- Genetic Testing Registry: Congenital myopathy 4B, autosomal recessive
- Genetic Testing Registry: Nemaline myopathy
- Genetic Testing Registry: Nemaline myopathy 10
- Genetic Testing Registry: Nemaline myopathy 2
- Genetic Testing Registry: Nemaline myopathy 5
- Genetic Testing Registry: Nemaline myopathy 6
- Genetic Testing Registry: Nemaline myopathy 7
- Genetic Testing Registry: Nemaline myopathy 8
- Genetic Testing Registry: Nemaline myopathy 9
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- CONGENITAL MYOPATHY 2A, TYPICAL, AUTOSOMAL DOMINANT; CMYP2A
- NEMALINE MYOPATHY 2; NEM2
- NEMALINE MYOPATHY 5A, AUTOSOMAL RECESSIVE, SEVERE INFANTILE; NEM5A
- NEMALINE MYOPATHY 7; NEM7
- NEMALINE MYOPATHY 6; NEM6
- CONGENITAL MYOPATHY 4B, AUTOSOMAL RECESSIVE; CMYP4B
- CONGENITAL MYOPATHY 23; CMYP23
- NEMALINE MYOPATHY 8; NEM8
- NEMALINE MYOPATHY 9; NEM9
- NEMALINE MYOPATHY 10; NEM10
Scientific Articles on PubMed
References
- Lehtokari VL, Pelin K, Sandbacka M, Ranta S, Donner K, Muntoni F, Sewry C, Angelini C, Bushby K, Van den Bergh P, Iannaccone S, Laing NG, Wallgren-Pettersson C. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat. 2006 Sep;27(9):946-56. doi: 10.1002/humu.20370. Citation on PubMed
- Nowak KJ, Sewry CA, Navarro C, Squier W, Reina C, Ricoy JR, Jayawant SS, Childs AM, Dobbie JA, Appleton RE, Mountford RC, Walker KR, Clement S, Barois A, Muntoni F, Romero NB, Laing NG. Nemaline myopathy caused by absence of alpha-skeletal muscle actin. Ann Neurol. 2007 Feb;61(2):175-84. doi: 10.1002/ana.21035. Citation on PubMed
- Ottenheijm CA, Hooijman P, DeChene ET, Stienen GJ, Beggs AH, Granzier H. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J Struct Biol. 2010 May;170(2):334-43. doi: 10.1016/j.jsb.2009.11.013. Epub 2009 Nov 26. Citation on PubMed or Free article on PubMed Central
- Romero NB, Sandaradura SA, Clarke NF. Recent advances in nemaline myopathy. Curr Opin Neurol. 2013 Oct;26(5):519-26. doi: 10.1097/WCO.0b013e328364d681. Citation on PubMed
- Ryan MM, Schnell C, Strickland CD, Shield LK, Morgan G, Iannaccone ST, Laing NG, Beggs AH, North KN. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol. 2001 Sep;50(3):312-20. doi: 10.1002/ana.1080. Citation on PubMed
- Wallgren-Pettersson C, Sewry CA, Nowak KJ, Laing NG. Nemaline myopathies. Semin Pediatr Neurol. 2011 Dec;18(4):230-8. doi: 10.1016/j.spen.2011.10.004. Citation on PubMed
- Yin X, Pu CQ, Wang Q, Liu JX, Mao YL. Clinical and pathological features of patients with nemaline myopathy. Mol Med Rep. 2014 Jul;10(1):175-82. doi: 10.3892/mmr.2014.2184. Epub 2014 Apr 24. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.