

Description
Naegeli-Franceschetti-Jadassohn syndrome/dermatopathia pigmentosa reticularis (NFJS/DPR) represents a rare type of ectodermal dysplasia, a group of about 150 conditions characterized by abnormal development of ectodermal tissues including the skin, hair, nails, teeth, and sweat glands. NFJS and DPR were originally described as separate conditions; however, because they have similar features and are caused by mutations in the same gene, they are now often considered forms of the same disorder.
Among the most common signs of NFJS/DPR is a net-like pattern of dark brown or gray skin coloring, known as reticulate hyperpigmentation. This darker pigmentation is seen most often on the neck, chest, and abdomen, although it can also occur in and around the eyes and mouth. Reticulate hyperpigmentation appears in infancy or early childhood. It may fade with age or persist throughout life.
NFJS/DPR also affects the skin on the hands and feet. The skin on the palms of the hands and soles of the feet often becomes thick, hard, and callused, a condition known as palmoplantar keratoderma. Some affected individuals also have blistering on their palms and soles. Their fingernails and toenails may be malformed, brittle, and either thicker or thinner than usual. Most affected individuals are missing the patterned ridges on the skin of the hands and feet, called dermatoglyphs, that are the basis for each person's unique fingerprints.
Additional features of NFJS/DPR can include a reduced ability to sweat (hypohidrosis) or excess sweating (hyperhidrosis) and dental abnormalities. Some affected individuals also have hair loss (alopecia) on the scalp, eyebrows, and underarms. The alopecia is described as noncicatricial because it does not leave scars (cicatrices).
Frequency
NFJS/DPR is a rare condition; its prevalence is unknown. Only a few affected families have been reported in the medical literature.
Causes
NFJS/DPR results from mutations in the KRT14 gene. This gene provides instructions for making a protein called keratin 14. Keratins are tough, fibrous proteins that provide strength and resiliency to the outer layer of the skin (the epidermis). Researchers believe that keratin 14 may also play a role in the formation of sweat glands and the development of dermatoglyphs.
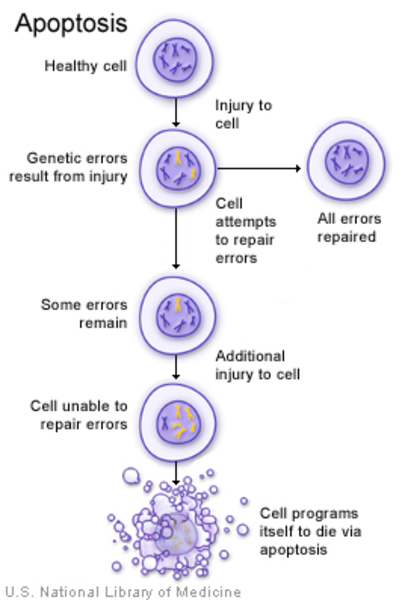
The KRT14 gene mutations that cause NFJS/DPR most likely reduce the amount of functional keratin 14 that is produced in cells. A shortage of this protein makes cells in the epidermis more likely to self-destruct (undergo apoptosis). The resulting loss of these cells alters the normal development and structure of ectodermal tissues, which likely underlies most of the skin and nail problems characteristic of NFJS/DPR. However, it is unclear how a shortage of keratin 14 is related to changes in skin pigmentation.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- DPR
- Franceschetti-Jadassohn syndrome
- Naegeli syndrome
- Naegeli-Franceschetti-Jadassohn syndrome
- NFJ syndrome
- NFJS
- NFJS/DPR
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Heimer WL 2nd, Brauner G, James WD. Dermatopathia pigmentosa reticularis: a report of a family demonstrating autosomal dominant inheritance. J Am Acad Dermatol. 1992 Feb;26(2 Pt 2):298-301. doi: 10.1016/0190-9622(92)70039-i. Citation on PubMed
- Itin PH, Lautenschlager S, Meyer R, Mevorah B, Rufli T. Natural history of the Naegeli-Franceschetti-Jadassohn syndrome and further delineation of its clinical manifestations. J Am Acad Dermatol. 1993 Jun;28(6):942-50. doi: 10.1016/0190-9622(93)70135-g. Citation on PubMed
- Lugassy J, Itin P, Ishida-Yamamoto A, Holland K, Huson S, Geiger D, Hennies HC, Indelman M, Bercovich D, Uitto J, Bergman R, McGrath JA, Richard G, Sprecher E. Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: two allelic ectodermal dysplasias caused by dominant mutations in KRT14. Am J Hum Genet. 2006 Oct;79(4):724-30. doi: 10.1086/507792. Epub 2006 Aug 25. Citation on PubMed or Free article on PubMed Central
- Lugassy J, McGrath JA, Itin P, Shemer R, Verbov J, Murphy HR, Ishida-Yamamoto A, Digiovanna JJ, Bercovich D, Karin N, Vitenshtein A, Uitto J, Bergman R, Richard G, Sprecher E. KRT14 haploinsufficiency results in increased susceptibility of keratinocytes to TNF-alpha-induced apoptosis and causes Naegeli-Franceschetti-Jadassohn syndrome. J Invest Dermatol. 2008 Jun;128(6):1517-24. doi: 10.1038/sj.jid.5701187. Epub 2007 Nov 29. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.