

Description
Mucolipidosis III gamma is a slowly progressive disorder that affects many parts of the body. Signs and symptoms of this condition typically appear around age 3.
Individuals with mucolipidosis III gamma grow slowly and have short stature. They also have stiff joints and dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. Many affected individuals develop low bone mineral density (osteoporosis), which weakens the bones and makes them prone to fracture. Osteoporosis and progressive joint problems in people with mucolipidosis III gamma also cause pain, which becomes more severe over time.
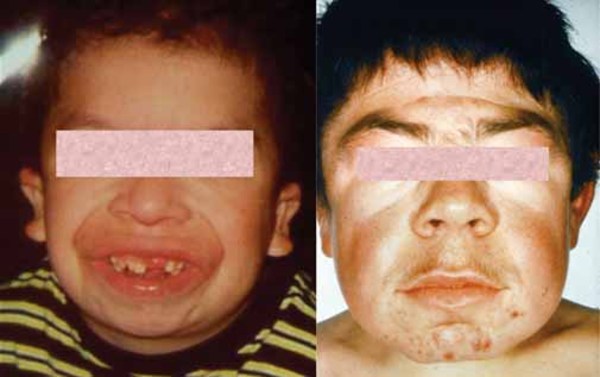
People with mucolipidosis III gamma often have heart valve abnormalities and mild clouding of the clear covering of the eye (cornea). Their facial features become slightly thickened or "coarse" as they get older. A small percentage of people with this condition have mild intellectual disability or learning problems. Individuals with mucolipidosis III gamma generally survive into adulthood, but they may have a shortened lifespan.

Frequency
Mucolipidosis III gamma is a rare disorder, although its exact prevalence is unknown. It is estimated to occur in about 1 in 100,000 to 400,000 individuals worldwide.
Causes
Mutations in the GNPTG gene cause mucolipidosis III gamma. This gene provides instructions for making one part (subunit) of an enzyme called GlcNAc-1-phosphotransferase. This enzyme helps prepare certain newly made enzymes for transport to lysosomes. Lysosomes are compartments within the cell that use digestive enzymes to break down large molecules into smaller ones that can be reused by cells. GlcNAc-1-phosphotransferase is involved in the process of attaching a molecule called mannose-6-phosphate (M6P) to specific digestive enzymes. Just as luggage is tagged at the airport to direct it to the correct destination, enzymes are often "tagged" after they are made so they get to where they are needed in the cell. M6P acts as a tag that indicates a digestive enzyme should be transported to the lysosome.

Mutations in the GNPTG gene that cause mucolipidosis III gamma result in reduced activity of GlcNAc-1-phosphotransferase. These mutations disrupt the tagging of digestive enzymes with M6P, which prevents many enzymes from reaching the lysosomes. Digestive enzymes that do not receive the M6P tag end up outside the cell, where they have increased activity. The shortage of digestive enzymes within lysosomes causes large molecules to accumulate there. Conditions that cause molecules to build up inside lysosomes, including mucolipidosis III gamma, are called lysosomal storage disorders. The signs and symptoms of mucolipidosis III gamma are most likely due to the shortage of digestive enzymes inside lysosomes and the effects these enzymes have outside the cell.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- ML IIIC
- Mucolipidosis III
- Mucolipidosis III, variant
- Mucolipidosis IIIC
- Mucolipidosis type III
- Pseudo-Hurler polydystrophy
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Cathey SS, Kudo M, Tiede S, Raas-Rothschild A, Braulke T, Beck M, Taylor HA, Canfield WM, Leroy JG, Neufeld EF, McKusick VA. Molecular order in mucolipidosis II and III nomenclature. Am J Med Genet A. 2008 Feb 15;146A(4):512-3. doi: 10.1002/ajmg.a.32193. No abstract available. Citation on PubMed
- Encarnacao M, Lacerda L, Costa R, Prata MJ, Coutinho MF, Ribeiro H, Lopes L, Pineda M, Ignatius J, Galvez H, Mustonen A, Vieira P, Lima MR, Alves S. Molecular analysis of the GNPTAB and GNPTG genes in 13 patients with mucolipidosis type II or type III - identification of eight novel mutations. Clin Genet. 2009 Jul;76(1):76-84. doi: 10.1111/j.1399-0004.2009.01185.x. Citation on PubMed
- Liu S, Zhang W, Shi H, Meng Y, Qiu Z. Three novel homozygous mutations in the GNPTG gene that cause mucolipidosis type III gamma. Gene. 2014 Feb 10;535(2):294-8. doi: 10.1016/j.gene.2013.11.010. Epub 2013 Dec 6. Citation on PubMed
- Persichetti E, Chuzhanova NA, Dardis A, Tappino B, Pohl S, Thomas NS, Rosano C, Balducci C, Paciotti S, Dominissini S, Montalvo AL, Sibilio M, Parini R, Rigoldi M, Di Rocco M, Parenti G, Orlacchio A, Bembi B, Cooper DN, Filocamo M, Beccari T. Identification and molecular characterization of six novel mutations in the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTG) gene in patients with mucolipidosis III gamma. Hum Mutat. 2009 Jun;30(6):978-84. doi: 10.1002/humu.20959. Citation on PubMed
- Pohl S, Tiede S, Castrichini M, Cantz M, Gieselmann V, Braulke T. Compensatory expression of human N-acetylglucosaminyl-1-phosphotransferase subunits in mucolipidosis type III gamma. Biochim Biophys Acta. 2009 Mar;1792(3):221-5. doi: 10.1016/j.bbadis.2009.01.009. Citation on PubMed
- Raas-Rothschild A, Bargal R, Goldman O, Ben-Asher E, Groener JE, Toutain A, Stemmer E, Ben-Neriah Z, Flusser H, Beemer FA, Penttinen M, Olender T, Rein AJ, Bach G, Zeigler M. Genomic organisation of the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTAG) and its mutations in mucolipidosis III. J Med Genet. 2004 Apr;41(4):e52. doi: 10.1136/jmg.2003.015222. No abstract available. Citation on PubMed or Free article on PubMed Central
- Raas-Rothschild A, Cormier-Daire V, Bao M, Genin E, Salomon R, Brewer K, Zeigler M, Mandel H, Toth S, Roe B, Munnich A, Canfield WM. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). J Clin Invest. 2000 Mar;105(5):673-81. doi: 10.1172/JCI5826. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.