

Description
Mucolipidosis II alpha/beta (also known as I-cell disease) is a progressively debilitating disorder that affects many parts of the body. Most affected individuals do not survive past early childhood.
At birth, children with mucolipidosis II alpha/beta are small and have weak muscle tone (hypotonia) and a weak cry. Affected individuals grow slowly after birth and usually stop growing during the second year of life. Development is delayed, particularly the development of speech and motor skills such as sitting and standing.
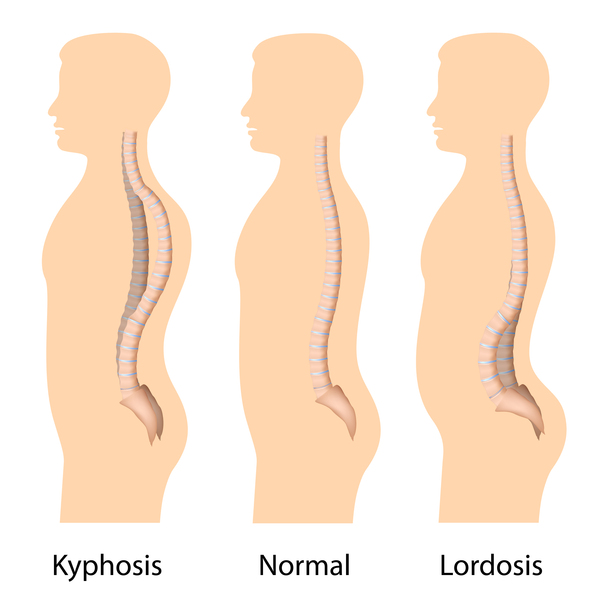
Children with mucolipidosis II alpha/beta typically have several bone abnormalities, many of which are present at birth. Affected individuals may have an abnormally rounded upper back (kyphosis), feet that are abnormally rotated (clubfeet), dislocated hips, unusually shaped long bones, and short hands and fingers. People with this condition also have joint deformities (contractures) that significantly affect mobility. Most children with mucolipidosis II alpha/beta do not develop the ability to walk independently. Affected individuals have dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray.

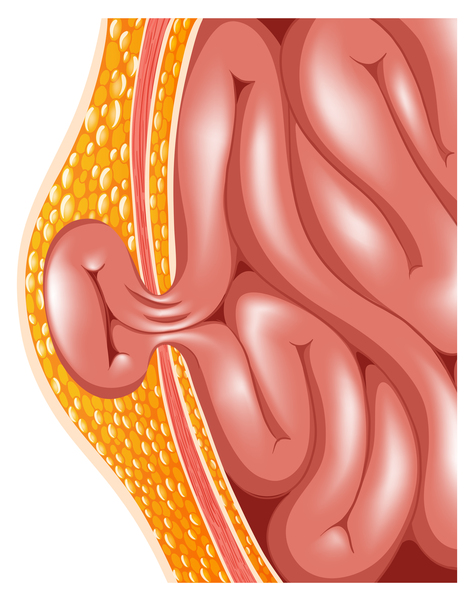
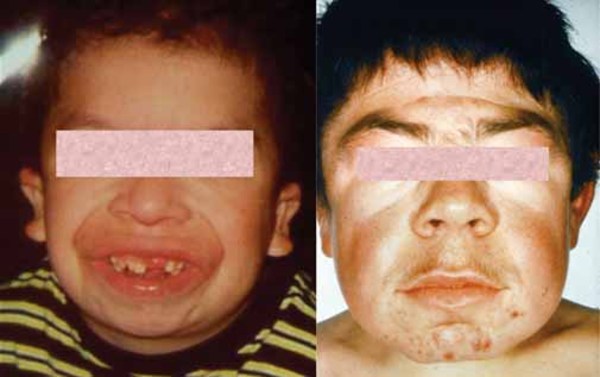
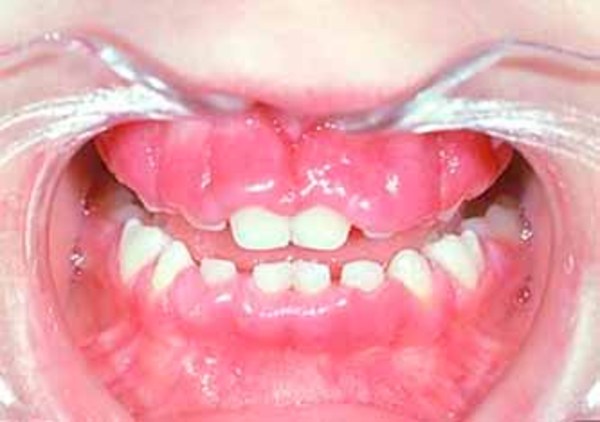
Other features of mucolipidosis II alpha/beta include a soft out-pouching around the belly-button (umbilical hernia) or lower abdomen (inguinal hernia), heart valve abnormalities, distinctive-looking facial features that are described as "coarse," and overgrowth of the gums (gingival hypertrophy). Vocal cords can stiffen, resulting in a hoarse voice. The airway is narrow, which can contribute to prolonged or recurrent respiratory infections. Affected individuals may also have recurrent ear infections, which can lead to hearing loss.
Frequency
Mucolipidosis II alpha/beta is a rare disorder, although its exact prevalence is unknown. It is estimated to occur in about 1 in 100,000 to 400,000 individuals worldwide.
Causes
Mutations in the GNPTAB gene cause mucolipidosis II alpha/beta. This gene provides instructions for making part of an enzyme called GlcNAc-1-phosphotransferase. This enzyme helps prepare certain newly made enzymes for transport to lysosomes. Lysosomes are compartments within the cell that use digestive enzymes to break down large molecules into smaller ones that can be reused by cells. GlcNAc-1-phosphotransferase is involved in the process of attaching a molecule called mannose-6-phosphate (M6P) to specific digestive enzymes. Just as luggage is tagged at the airport to direct it to the correct destination, enzymes are often "tagged" after they are made so they get to where they are needed in the cell. M6P acts as a tag that indicates a digestive enzyme should be transported to the lysosome.

Mutations in the GNPTAB gene that cause mucolipidosis II alpha/beta prevent the production of any functional GlcNAc-1-phosphotransferase. Without this enzyme, digestive enzymes cannot be tagged with M6P and transported to lysosomes. Instead, they end up outside the cell and have increased digestive activity. The lack of digestive enzymes within lysosomes causes large molecules to accumulate there. Conditions that cause molecules to build up inside lysosomes, including mucolipidosis II alpha/beta, are called lysosomal storage disorders. The signs and symptoms of mucolipidosis II alpha/beta are most likely caused by the lack of digestive enzymes within lysosomes and the effects these enzymes have outside the cell.
Mutations in the GNPTAB gene can also cause a similar but milder disorder called mucolipidosis III alpha/beta. Instead of preventing the production of any enzyme, these mutations reduce the activity of GlcNAc-1-phosphotransferase. Mucolipidosis III alpha/beta and mucolipidosis II alpha/beta represent two ends of a spectrum of disease severity.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- I-cell disease
- Inclusion cell disease
- MLII
- Mucolipidosis II
- Mucolipidosis type II
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bargal R, Zeigler M, Abu-Libdeh B, Zuri V, Mandel H, Ben Neriah Z, Stewart F, Elcioglu N, Hindi T, Le Merrer M, Bach G, Raas-Rothschild A. When Mucolipidosis III meets Mucolipidosis II: GNPTA gene mutations in 24 patients. Mol Genet Metab. 2006 Aug;88(4):359-63. doi: 10.1016/j.ymgme.2006.03.003. Epub 2006 Apr 21. Erratum In: Mol Genet Metab. 2007 Jul;91(3):299. Citation on PubMed
- Braulke T, Pohl S, Storch S. Molecular analysis of the GlcNac-1-phosphotransferase. J Inherit Metab Dis. 2008 Apr;31(2):253-7. doi: 10.1007/s10545-008-0862-5. Epub 2008 Apr 15. Citation on PubMed
- Cathey SS, Kudo M, Tiede S, Raas-Rothschild A, Braulke T, Beck M, Taylor HA, Canfield WM, Leroy JG, Neufeld EF, McKusick VA. Molecular order in mucolipidosis II and III nomenclature. Am J Med Genet A. 2008 Feb 15;146A(4):512-3. doi: 10.1002/ajmg.a.32193. No abstract available. Citation on PubMed
- Cathey SS, Leroy JG, Wood T, Eaves K, Simensen RJ, Kudo M, Stevenson RE, Friez MJ. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. J Med Genet. 2010 Jan;47(1):38-48. doi: 10.1136/jmg.2009.067736. Epub 2009 Jul 16. Citation on PubMed or Free article on PubMed Central
- Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase alpha / beta -subunits precursor gene. Am J Hum Genet. 2006 Mar;78(3):451-63. doi: 10.1086/500849. Epub 2006 Jan 24. Citation on PubMed or Free article on PubMed Central
- Leroy JG, Cathey SS, Friez MJ. GNPTAB-Related Disorders. 2008 Aug 26 [updated 2019 Aug 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1828/ Citation on PubMed
- Otomo T, Higaki K, Nanba E, Ozono K, Sakai N. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J Biol Chem. 2011 Oct 7;286(40):35283-90. doi: 10.1074/jbc.M111.267930. Epub 2011 Aug 16. Citation on PubMed or Free article on PubMed Central
- Otomo T, Muramatsu T, Yorifuji T, Okuyama T, Nakabayashi H, Fukao T, Ohura T, Yoshino M, Tanaka A, Okamoto N, Inui K, Ozono K, Sakai N. Mucolipidosis II and III alpha/beta: mutation analysis of 40 Japanese patients showed genotype-phenotype correlation. J Hum Genet. 2009 Mar;54(3):145-51. doi: 10.1038/jhg.2009.3. Epub 2009 Feb 6. Citation on PubMed
- Plante M, Claveau S, Lepage P, Lavoie EM, Brunet S, Roquis D, Morin C, Vezina H, Laprise C. Mucolipidosis II: a single causal mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTAB) in a French Canadian founder population. Clin Genet. 2008 Mar;73(3):236-44. doi: 10.1111/j.1399-0004.2007.00954.x. Epub 2008 Jan 7. Citation on PubMed
- Saul RA, Proud V, Taylor HA, Leroy JG, Spranger J. Prenatal mucolipidosis type II (I-cell disease) can present as Pacman dysplasia. Am J Med Genet A. 2005 Jun 15;135(3):328-32. doi: 10.1002/ajmg.a.30716. Citation on PubMed
- Takanashi J, Hayashi M, Yuasa S, Satoh H, Terada H. Hypoyelination in I-cell disease; MRI, MR spectroscopy and neuropathological correlation. Brain Dev. 2012 Oct;34(9):780-3. doi: 10.1016/j.braindev.2011.12.013. Epub 2012 Jan 24. Citation on PubMed
- Tiede S, Storch S, Lubke T, Henrissat B, Bargal R, Raas-Rothschild A, Braulke T. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat Med. 2005 Oct;11(10):1109-12. doi: 10.1038/nm1305. Epub 2005 Oct 2. Citation on PubMed
- Wilcox WR, Wenger DA, Lachman RS, Rimoin DL. Distinguishing Pacman dysplasia from mucolipidosis II: comment on Saul et al. [2005]. Am J Med Genet A. 2005 Jun 15;135(3):333. doi: 10.1002/ajmg.a.30717. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.