

Description
Miller-Dieker syndrome is a condition characterized by a pattern of abnormal brain development known as lissencephaly. Normally the exterior of the brain (cerebral cortex) is multi-layered with folds and grooves. People with lissencephaly have an abnormally smooth brain with fewer folds and grooves. These brain malformations cause severe intellectual disability, developmental delay, seizures, abnormal muscle stiffness (spasticity), weak muscle tone (hypotonia), and feeding difficulties. Seizures usually begin before six months of age, and some occur from birth. Typically, the smoother the surface of the brain is, the more severe the associated symptoms are.

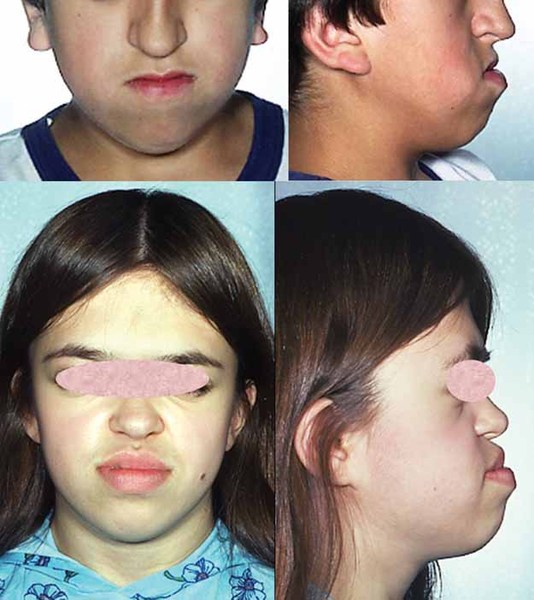
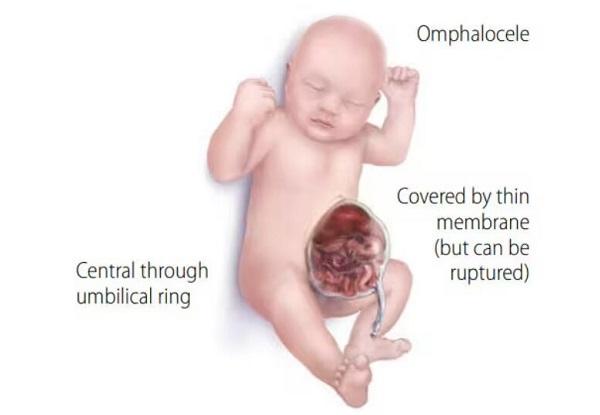
In addition to lissencephaly, people with Miller-Dieker syndrome tend to have distinctive facial features that include a prominent forehead; a sunken appearance in the middle of the face (midface hypoplasia); a small, upturned nose; low-set and abnormally shaped ears; a small jaw; and a thick upper lip. Some individuals with this condition also grow more slowly than other children. Rarely, affected individuals will have heart or kidney malformations or an opening in the wall of the abdomen (an omphalocele) that allows the abdominal organs to protrude through the navel. People with Miller-Dieker syndrome may also have life-threatening breathing problems. Most individuals with this condition do not survive beyond childhood.

Frequency
Miller-Dieker syndrome appears to be a rare disorder, although its prevalence is unknown.
Causes
Miller-Dieker syndrome is caused by a deletion of genetic material near the end of the short (p) arm of chromosome 17. The signs and symptoms of Miller-Dieker syndrome are probably related to the loss of multiple genes in this region. The size of the deletion varies among affected individuals.
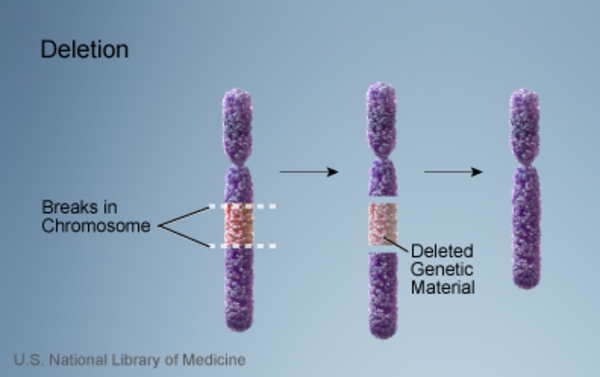
Researchers are working to identify all of the genes that contribute to the features of Miller-Dieker syndrome. They have determined that the loss of a particular gene on chromosome 17, PAFAH1B1, is responsible for the syndrome's characteristic sign of lissencephaly. The loss of another gene, YWHAE, in the same region of chromosome 17 increases the severity of the lissencephaly in people with Miller-Dieker syndrome. Additional genes in the deleted region probably contribute to the varied features of Miller-Dieker syndrome.
Inheritance
Most cases of Miller-Dieker syndrome are not inherited. The deletion occurs most often as a random event during the formation of reproductive cells (eggs or sperm) or in early fetal development. Affected people typically have no history of the disorder in their family.
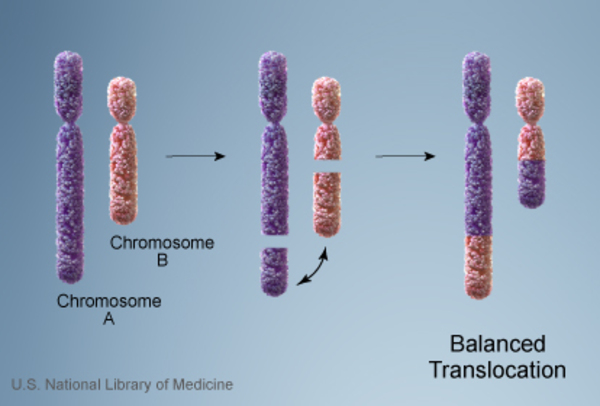
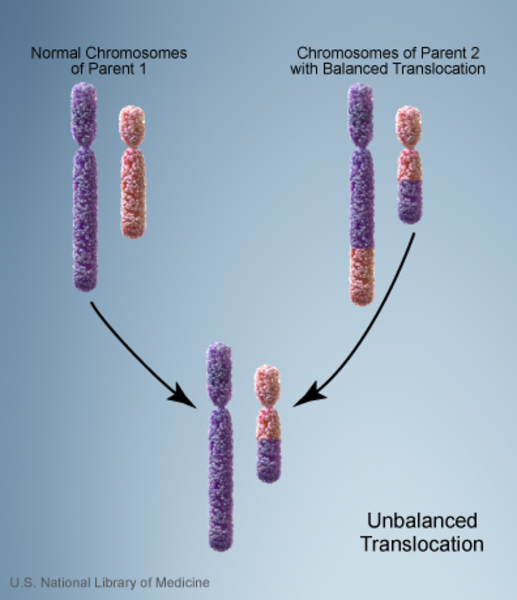
When Miller-Dieker syndrome is inherited, its inheritance pattern is considered autosomal dominant because a deletion in one copy of chromosome 17 in each cell is sufficient to cause the condition. About 12 percent of people with Miller-Dieker syndrome inherit a chromosome abnormality from an unaffected parent. In these cases, the parent carries a chromosomal rearrangement called a balanced translocation, in which no genetic material is gained or lost. Balanced translocations usually do not cause any health problems; however, they can become unbalanced as they are passed to the next generation. Children who inherit an unbalanced translocation can have a chromosomal rearrangement with extra or missing genetic material. Individuals with Miller-Dieker syndrome who inherit an unbalanced translocation are missing genetic material from the short arm of chromosome 17, which results in the health problems characteristic of this disorder.


Other Names for This Condition
- Classical lissencephaly syndrome
- MDS
- Miller-Dieker lissencephaly syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Allanson JE, Ledbetter DH, Dobyns WB. Classical lissencephaly syndromes: does the face reflect the brain? J Med Genet. 1998 Nov;35(11):920-3. doi: 10.1136/jmg.35.11.920. Citation on PubMed or Free article on PubMed Central
- Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, Martin CL, Allanson J, Pilz DT, Olney AH, Mutchinick OM, Hirotsune S, Wynshaw-Boris A, Dobyns WB, Ledbetter DH. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003 Apr;72(4):918-30. doi: 10.1086/374320. Epub 2003 Mar 5. Citation on PubMed or Free article on PubMed Central
- Dobyns WB, Curry CJ, Hoyme HE, Turlington L, Ledbetter DH. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991 Mar;48(3):584-94. Citation on PubMed or Free article on PubMed Central
- Nagamani SC, Zhang F, Shchelochkov OA, Bi W, Ou Z, Scaglia F, Probst FJ, Shinawi M, Eng C, Hunter JV, Sparagana S, Lagoe E, Fong CT, Pearson M, Doco-Fenzy M, Landais E, Mozelle M, Chinault AC, Patel A, Bacino CA, Sahoo T, Kang SH, Cheung SW, Lupski JR, Stankiewicz P. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J Med Genet. 2009 Dec;46(12):825-33. doi: 10.1136/jmg.2009.067637. Epub 2009 Jul 6. Citation on PubMed
- Spalice A, Parisi P, Nicita F, Pizzardi G, Del Balzo F, Iannetti P. Neuronal migration disorders: clinical, neuroradiologic and genetics aspects. Acta Paediatr. 2009 Mar;98(3):421-33. doi: 10.1111/j.1651-2227.2008.01160.x. Epub 2008 Dec 16. Citation on PubMed
- Sweeney KJ, Clark GD, Prokscha A, Dobyns WB, Eichele G. Lissencephaly associated mutations suggest a requirement for the PAFAH1B heterotrimeric complex in brain development. Mech Dev. 2000 Apr;92(2):263-71. doi: 10.1016/s0925-4773(00)00242-2. Citation on PubMed
- Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003 Jul;34(3):274-85. doi: 10.1038/ng1169. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.