

Description
Microcephalic osteodysplastic primordial dwarfism type II (MOPDII) is a condition characterized by short stature (dwarfism) with other skeletal abnormalities (osteodysplasia) and an unusually small head size (microcephaly). The growth problems in MOPDII are primordial, meaning they begin before birth, with affected individuals showing slow prenatal growth (intrauterine growth retardation). After birth, affected individuals continue to grow at a very slow rate. The final adult height of people with this condition ranges from 20 inches to 40 inches. Other skeletal abnormalities in MOPDII include abnormal development of the hip joints (hip dysplasia), thinning of the bones in the arms and legs, an abnormal side-to-side curvature of the spine (scoliosis), and shortened wrist bones. In people with MOPDII head growth slows over time; affected individuals have an adult brain size comparable to that of a 3-month-old infant. However, intellectual development is typically normal.

People with this condition have a high-pitched, nasal voice and some have a narrowing of the voicebox (subglottic stenosis). Facial features characteristic of MOPDII include a prominent nose, full cheeks, a long midface, and a small jaw. Other signs and symptoms seen in some people with MOPDII include small teeth (microdontia) and farsightedness. Over time, affected individuals may develop areas of abnormally light or dark skin coloring (pigmentation).
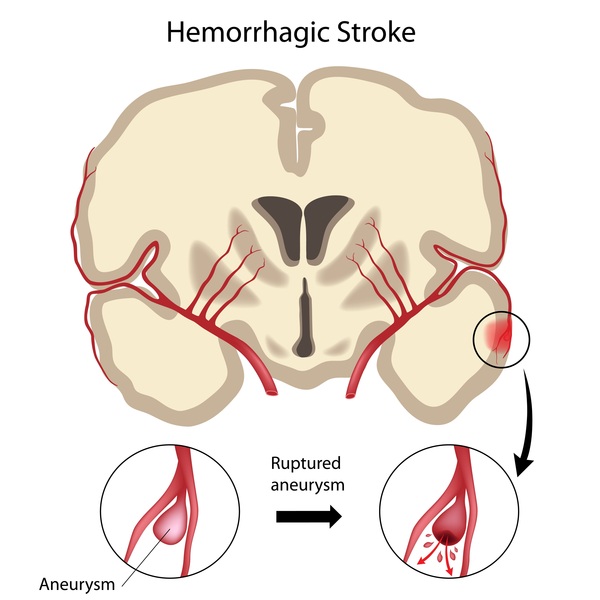
Many individuals with MOPDII have blood vessel abnormalities. For example, some affected individuals develop a bulge in one of the blood vessels at the center of the brain (intracranial aneurysm). These aneurysms are dangerous because they can burst, causing bleeding within the brain. Some affected individuals have Moyamoya disease, in which arteries at the base of the brain are narrowed, leading to restricted blood flow. These vascular abnormalities are often treatable, though they increase the risk of stroke and reduce the life expectancy of affected individuals.
Frequency
MOPDII appears to be a rare condition, although its prevalence is unknown.
Causes
Mutations in the PCNT gene cause MOPDII. The PCNT gene provides instructions for making a protein called pericentrin. Within cells, this protein is located in structures called centrosomes. Centrosomes play a role in cell division and the assembly of microtubules. Microtubules are fibers that help cells maintain their shape, assist in the process of cell division, and are essential for the transport of materials within cells. Pericentrin acts as an anchoring protein, securing other proteins to the centrosome. Through its interactions with these proteins, pericentrin plays a role in regulation of the cell cycle, which is the cell's way of replicating itself in an organized, step-by-step fashion.
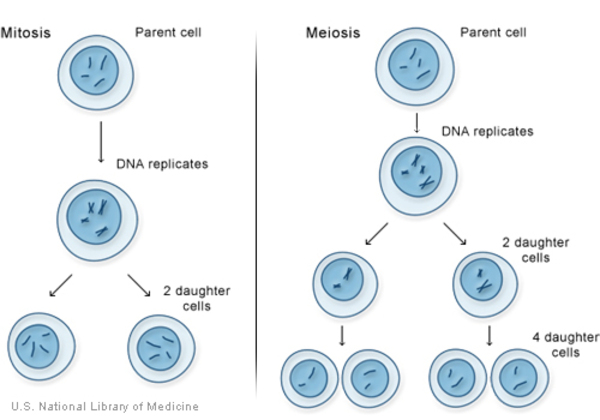
PCNT gene mutations lead to the production of a nonfunctional pericentrin protein that cannot anchor other proteins to the centrosome. As a result, centrosomes cannot properly assemble microtubules, leading to disruption of the cell cycle and cell division. Impaired cell division causes a reduction in cell production, while disruption of the cell cycle can lead to cell death. This overall reduction in the number of cells leads to short bones, microcephaly, and the other signs and symptoms of MOPDII.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Majewski osteodysplastic primordial dwarfism type II
- MOPD2
- MOPDII
- Osteodysplastic primordial dwarfism type II
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bober MB, Khan N, Kaplan J, Lewis K, Feinstein JA, Scott CI Jr, Steinberg GK. Majewski osteodysplastic primordial dwarfism type II (MOPD II): expanding the vascular phenotype. Am J Med Genet A. 2010 Apr;152A(4):960-5. doi: 10.1002/ajmg.a.33252. Citation on PubMed
- Hall JG, Flora C, Scott CI Jr, Pauli RM, Tanaka KI. Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. Am J Med Genet A. 2004 Sep 15;130A(1):55-72. doi: 10.1002/ajmg.a.30203. Citation on PubMed
- Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, Zweier C, Brunner HG, Becker K, Curry CJ, Dallapiccola B, Devriendt K, Dorfler A, Kinning E, Megarbane A, Meinecke P, Semple RK, Spranger S, Toutain A, Trembath RC, Voss E, Wilson L, Hennekam R, de Zegher F, Dorr HG, Reis A. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008 Feb 8;319(5864):816-9. doi: 10.1126/science.1151174. Epub 2008 Jan 3. Citation on PubMed
- Willems M, Genevieve D, Borck G, Baumann C, Baujat G, Bieth E, Edery P, Farra C, Gerard M, Heron D, Leheup B, Le Merrer M, Lyonnet S, Martin-Coignard D, Mathieu M, Thauvin-Robinet C, Verloes A, Colleaux L, Munnich A, Cormier-Daire V. Molecular analysis of pericentrin gene (PCNT) in a series of 24 Seckel/microcephalic osteodysplastic primordial dwarfism type II (MOPD II) families. J Med Genet. 2010 Dec;47(12):797-802. doi: 10.1136/jmg.2009.067298. Epub 2009 Jul 29. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.