

Description
Manitoba oculotrichoanal syndrome is a condition involving several characteristic physical features, particularly affecting the eyes (oculo-), hair (tricho-), and anus (-anal).
People with Manitoba oculotrichoanal syndrome have widely spaced eyes (hypertelorism). They may also have other eye abnormalities including small eyes (microphthalmia), a notched or partially absent upper eyelid (upper eyelid coloboma), eyelids that are attached to the front surface of the eye (corneopalpebral synechiae), or eyes that are completely covered by skin and usually malformed (cryptophthalmos). These abnormalities may affect one or both eyes.

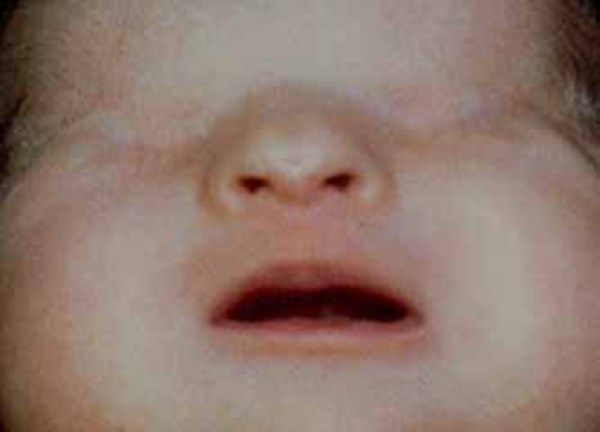
Individuals with Manitoba oculotrichoanal syndrome usually have abnormalities of the front hairline, such as hair growth extending from the temple to the eye on one or both sides of the face. One or both eyebrows may be completely or partially missing. Most people with this disorder also have a wide nose with a notched tip; in some cases this notch extends up from the tip so that the nose appears to be divided into two halves (bifid nose).
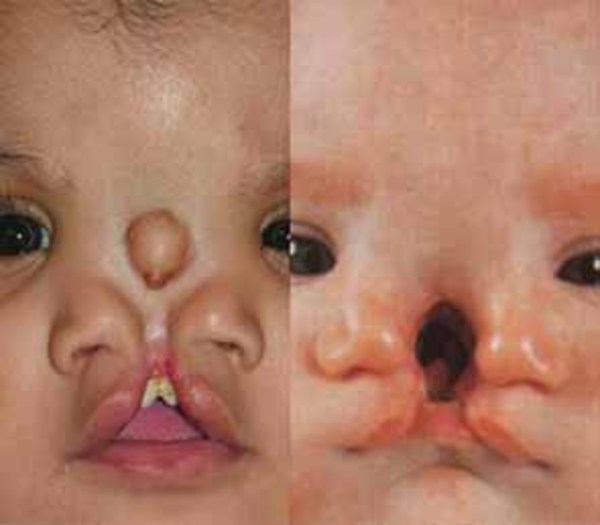
About 20 percent of people with Manitoba oculotrichoanal syndrome have defects in the abdominal wall, such as a soft out-pouching around the belly-button (an umbilical hernia) or an opening in the wall of the abdomen (an omphalocele) that allows the abdominal organs to protrude through the navel. Another characteristic feature of Manitoba oculotrichoanal syndrome is a narrow anus (anal stenosis) or an anal opening farther forward than usual. Umbilical wall defects or anal malformations may require surgical correction. Some affected individuals also have malformations of the kidneys.
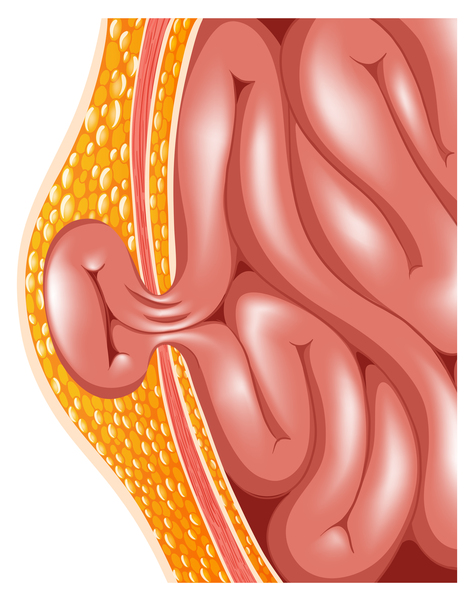
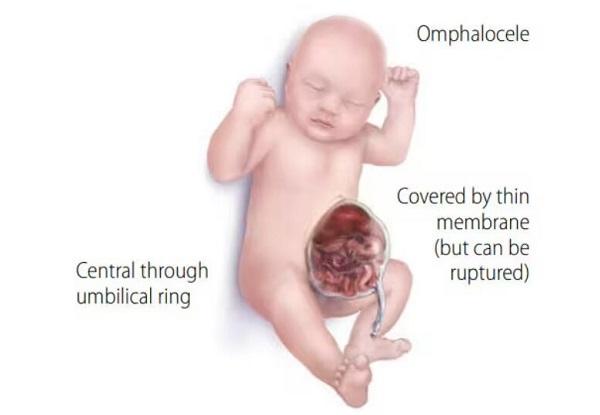
The severity of the features of Manitoba oculotrichoanal syndrome may vary even within the same family. With appropriate treatment, affected individuals generally have normal growth and development, intelligence, and life expectancy.
Frequency
Manitoba oculotrichoanal syndrome is estimated to occur in 2 to 6 in 1,000 people in a small isolated Ojibway-Cree community in northern Manitoba, Canada. Although this region has the highest incidence of the condition, it has also been diagnosed in a few people from other parts of the world.
Causes
Manitoba oculotrichoanal syndrome is caused by mutations in the FREM1 gene.
The FREM1 gene provides instructions for making a protein that is involved in the formation and organization of basement membranes, which are thin, sheet-like structures that separate and support cells in many tissues.
The FREM1 protein is one of a group of proteins, including proteins called FRAS1 and FREM2, that interact during embryonic development as components of basement membranes. Basement membranes help anchor layers of cells lining the surfaces and cavities of the body (epithelial cells) to other embryonic tissues, including those that give rise to connective tissues such as skin and cartilage.
The FREM1 gene mutations that have been identified in people with Manitoba oculotrichoanal syndrome delete genetic material from the FREM1 gene or result in a premature stop signal that leads to an abnormally short FREM1 protein. These mutations most likely result in a nonfunctional protein.
Absence of functional FREM1 protein interferes with its role in embryonic basement membrane development and may also affect the location, stability, or function of the FRAS1 and FREM2 proteins. The features of Manitoba oculotrichoanal syndrome may result from the failure of neighboring embryonic tissues to fuse properly due to impairment of the basement membranes' anchoring function.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Marles Greenberg Persaud syndrome
- Marles syndrome
- Marles-Greenberg-Persaud syndrome
- MOTA
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Li C, Marles SL, Greenberg CR, Chodirker BN, van de Kamp J, Slavotinek A, Chudley AE. Manitoba Oculotrichoanal (MOTA) syndrome: report of eight new cases. Am J Med Genet A. 2007 Apr 15;143A(8):853-7. doi: 10.1002/ajmg.a.31446. Citation on PubMed
- Li C, Slavotinek A. FREM1 Autosomal Recessive Disorders. 2008 Jul 9 [updated 2019 May 9]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1728/ Citation on PubMed
- Marles SL, Greenberg CR, Persaud TV, Shuckett EP, Chudley AE. New familial syndrome of unilateral upper eyelid coloboma, aberrant anterior hairline pattern, and anal anomalies in Manitoba Indians. Am J Med Genet. 1992 Apr 1;42(6):793-9. doi: 10.1002/ajmg.1320420609. Citation on PubMed
- Slavotinek AM, Baranzini SE, Schanze D, Labelle-Dumais C, Short KM, Chao R, Yahyavi M, Bijlsma EK, Chu C, Musone S, Wheatley A, Kwok PY, Marles S, Fryns JP, Maga AM, Hassan MG, Gould DB, Madireddy L, Li C, Cox TC, Smyth I, Chudley AE, Zenker M. Manitoba-oculo-tricho-anal (MOTA) syndrome is caused by mutations in FREM1. J Med Genet. 2011 Jun;48(6):375-82. doi: 10.1136/jmg.2011.089631. Epub 2011 Apr 20. Citation on PubMed or Free article on PubMed Central
- Yeung A, Amor D, Savarirayan R. Familial upper eyelid coloboma with ipsilateral anterior hairline abnormality: two new reports of MOTA syndrome. Am J Med Genet A. 2009 Feb 15;149A(4):767-9. doi: 10.1002/ajmg.a.32743. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.