

Description
Mabry syndrome is a condition characterized by intellectual disability, distinctive facial features, increased levels of an enzyme called alkaline phosphatase in the blood (hyperphosphatasia), and other signs and symptoms.
People with Mabry syndrome have intellectual disability that is often moderate to severe. They typically have little to no speech development and are delayed in the development of motor skills (such as sitting, crawling, and walking). Many affected individuals have low muscle tone (hypotonia) and develop recurrent seizures (epilepsy) in early childhood. Seizures are usually the generalized tonic-clonic type, which involve muscle rigidity, convulsions, and loss of consciousness.
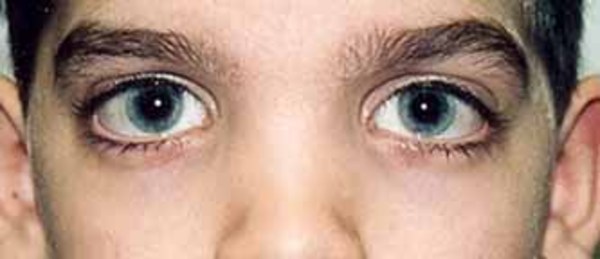
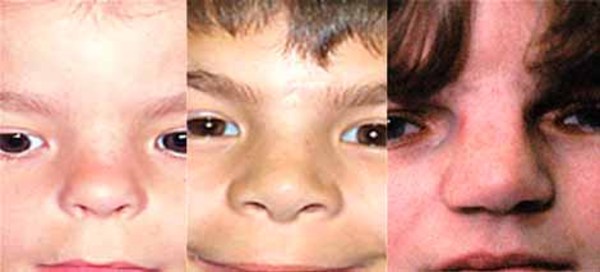
Individuals with Mabry syndrome have distinctive facial features that include wide-set eyes (hypertelorism), long openings of the eyelids (long palpebral fissures), a nose with a broad bridge and a rounded tip, downturned corners of the mouth, and a thin upper lip. These facial features usually become less pronounced over time.


Hyperphosphatasia begins within the first year of life in people with Mabry syndrome. There are many different types of alkaline phosphatase found in tissues; the type that is increased in Mabry syndrome is called the tissue non-specific type and is found throughout the body. In affected individuals, alkaline phosphatase levels in the blood are usually increased by one to two times the normal amount, but can be up to 20 times higher than normal. The elevated enzyme levels remain relatively stable over a person's lifetime. Hyperphosphatasia appears to cause no negative health effects, but this finding can help health professionals diagnose Mabry syndrome.
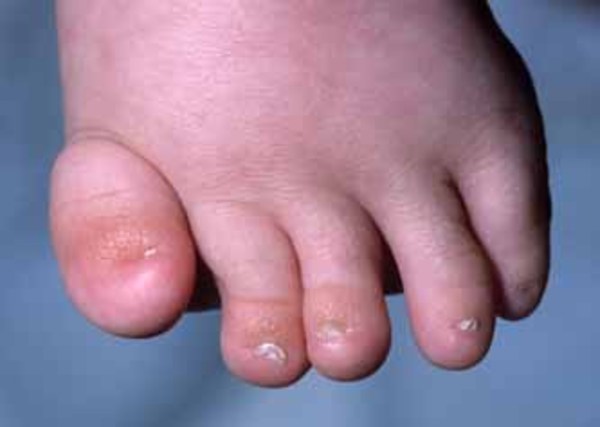
Another common feature of Mabry syndrome is shortened bones at the ends of fingers (brachytelephalangy), which can be seen on x-ray imaging. Underdeveloped fingernails (nail hypoplasia) may also occur. Sometimes, individuals with Mabry syndrome have abnormalities of the digestive system, including narrowing or blockage of the anus (anal stenosis or anal atresia) or Hirschsprung disease, a disorder that causes severe constipation or blockage of the intestine. Rarely, affected individuals experience hearing loss.
The signs and symptoms of Mabry syndrome vary among affected individuals. Those who are least severely affected have only intellectual disability and hyperphosphatasia, without distinctive facial features or the other health problems listed above.
Frequency
Mabry syndrome is likely a rare condition, but its prevalence is unknown. More than 20 cases have been described in the scientific literature.
Causes
Mutations in the PIGV, PIGO, or PGAP2 gene cause Mabry syndrome. These genes are all involved in the production (synthesis) of a molecule called a glycosylphosphosphatidylinositol (GPI) anchor. This molecule is synthesized in a series of steps. It then attaches (binds) to various proteins and binds them to the outer surface of the cell membrane, ensuring that they are available when needed. Alkaline phosphatase is an example of a protein that is bound to the cell membrane by a GPI anchor.
The proteins produced from the PIGV and PIGO genes are involved in piecing together the GPI anchor. After the complete GPI anchor is attached to a protein, the protein produced from the PGAP2 gene adjusts the anchor to enhance the anchor's ability to bind to the cell membrane.
Mutations in the PIGV, PIGO, or PGAP2 gene result in the production of an incomplete GPI anchor that cannot attach to proteins or to cell membranes. Proteins lacking a functional GPI anchor cannot bind to the cell membrane and are instead released from the cell. The release of non-GPI anchored alkaline phosphatase elevates the amount of this protein in the blood, causing hyperphosphatasia in people with Mabry syndrome. It is unclear how gene mutations lead to the other features of Mabry syndrome, but these signs and symptoms are likely due to a lack of proper GPI anchoring of proteins.
PIGV gene mutations are the most frequent cause of Mabry syndrome, accounting for approximately half of all cases. Mutations in the PIGO and PGAP2 genes are responsible for a small proportion of Mabry syndrome. The remaining affected individuals do not have an identified mutation in any of these three genes; the cause of the condition in these individuals is unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Hyperphosphatasia with mental retardation syndrome
- Hyperphosphatasia with seizures and neurologic deficit
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Hyperphosphatasia with intellectual disability syndrome 1

- Genetic Testing Registry: Hyperphosphatasia with intellectual disability syndrome 2
- Genetic Testing Registry: Hyperphosphatasia with intellectual disability syndrome 3
- Genetic Testing Registry: Hyperphosphatasia-intellectual disability syndrome
Patient Support and Advocacy Resources
Scientific Articles on PubMed
References
- Hansen L, Tawamie H, Murakami Y, Mang Y, ur Rehman S, Buchert R, Schaffer S, Muhammad S, Bak M, Nothen MM, Bennett EP, Maeda Y, Aigner M, Reis A, Kinoshita T, Tommerup N, Baig SM, Abou Jamra R. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am J Hum Genet. 2013 Apr 4;92(4):575-83. doi: 10.1016/j.ajhg.2013.03.008. Citation on PubMed or Free article on PubMed Central
- Horn D, Krawitz P, Mannhardt A, Korenke GC, Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: expanded clinical spectrum. Am J Med Genet A. 2011 Aug;155A(8):1917-22. doi: 10.1002/ajmg.a.34102. Epub 2011 Jul 7. Citation on PubMed
- Horn D, Schottmann G, Meinecke P. Hyperphosphatasia with mental retardation, brachytelephalangy, and a distinct facial gestalt: Delineation of a recognizable syndrome. Eur J Med Genet. 2010 Mar-Apr;53(2):85-8. doi: 10.1016/j.ejmg.2010.01.002. Epub 2010 Jan 18. Citation on PubMed
- Krawitz PM, Murakami Y, Hecht J, Kruger U, Holder SE, Mortier GR, Delle Chiaie B, De Baere E, Thompson MD, Roscioli T, Kielbasa S, Kinoshita T, Mundlos S, Robinson PN, Horn D. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet. 2012 Jul 13;91(1):146-51. doi: 10.1016/j.ajhg.2012.05.004. Epub 2012 Jun 7. Citation on PubMed or Free article on PubMed Central
- Krawitz PM, Murakami Y, Riess A, Hietala M, Kruger U, Zhu N, Kinoshita T, Mundlos S, Hecht J, Robinson PN, Horn D. PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am J Hum Genet. 2013 Apr 4;92(4):584-9. doi: 10.1016/j.ajhg.2013.03.011. Citation on PubMed or Free article on PubMed Central
- Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C, Stephani F, Kinoshita T, Murakami Y, Bauer S, Isau M, Fischer A, Dahl A, Kerick M, Hecht J, Kohler S, Jager M, Grunhagen J, de Condor BJ, Doelken S, Brunner HG, Meinecke P, Passarge E, Thompson MD, Cole DE, Horn D, Roscioli T, Mundlos S, Robinson PN. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010 Oct;42(10):827-9. doi: 10.1038/ng.653. Epub 2010 Aug 29. Citation on PubMed
- Murakami Y, Kanzawa N, Saito K, Krawitz PM, Mundlos S, Robinson PN, Karadimitris A, Maeda Y, Kinoshita T. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J Biol Chem. 2012 Feb 24;287(9):6318-25. doi: 10.1074/jbc.M111.331090. Epub 2012 Jan 6. Citation on PubMed or Free article on PubMed Central
- Thompson MD, Nezarati MM, Gillessen-Kaesbach G, Meinecke P, Mendoza-Londono R, Mornet E, Brun-Heath I, Squarcioni CP, Legeai-Mallet L, Munnich A, Cole DE. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am J Med Genet A. 2010 Jul;152A(7):1661-9. doi: 10.1002/ajmg.a.33438. Erratum In: Am J Med Genet A. 2011 May;155A(5):1215. Mendoza, Roberto [corrected to Mendoza-Londono, Roberto]. Citation on PubMed
- Thompson MD, Roscioli T, Marcelis C, Nezarati MM, Stolte-Dijkstra I, Sharom FJ, Lu P, Phillips JA, Sweeney E, Robinson PN, Krawitz P, Yntema HG, Andrade DM, Brunner HG, Cole DE. Phenotypic variability in hyperphosphatasia with seizures and neurologic deficit (Mabry syndrome). Am J Med Genet A. 2012 Mar;158A(3):553-8. doi: 10.1002/ajmg.a.35202. Epub 2012 Feb 7. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.