

Description
Lateral meningocele syndrome is a disorder that affects the nervous system, the bones and muscles, and other body systems. The condition is characterized by abnormalities known as lateral meningoceles. Lateral meningoceles are protrusions of the membranes surrounding the spinal cord (known as the meninges) through gaps in the bones of the spine (vertebrae). The protrusions are most common and typically larger in the lower spine.
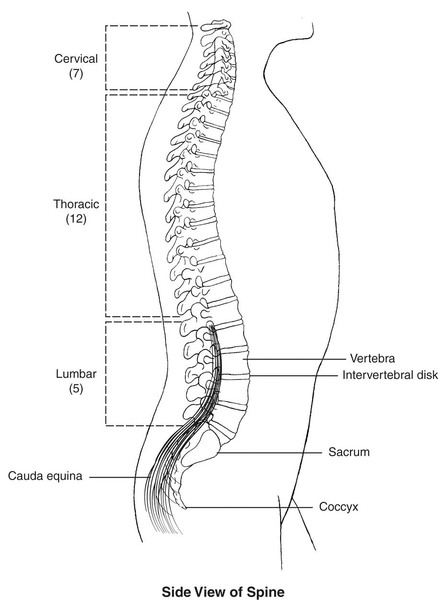
The meningoceles associated with this disorder may damage the nerves that spread from the spine to the rest of the body. Damage to the nerves that control bladder function, a condition called neurogenic bladder, causes affected individuals to have progressive difficulty controlling the flow of urine. Prickling or tingling sensations (paresthesias), progressive stiffness and weakness in the legs (paraparesis), and back pain can also occur. Delayed development of motor skills in infancy, such as sitting and crawling, often occurs in this disorder; intelligence is usually unaffected.
Other features of lateral meningocele syndrome can include low muscle tone (hypotonia) during infancy, decreased muscle bulk, loose (hyperextensible) joints that can lead to dislocations, and protrusion of organs through gaps in muscles (hernias). Spinal abnormalities are also common, including side-to-side curvature of the spine (scoliosis), abnormal joining (fusion) of two or more vertebrae, and vertebrae that are unusually shaped (scalloped).


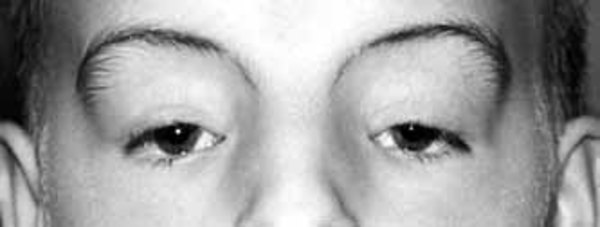
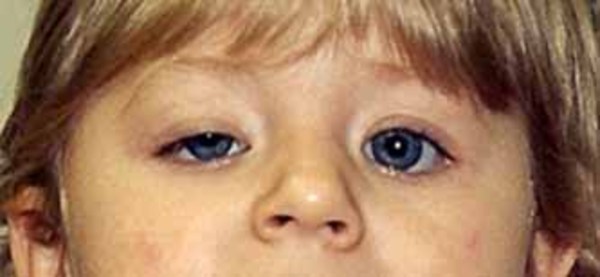
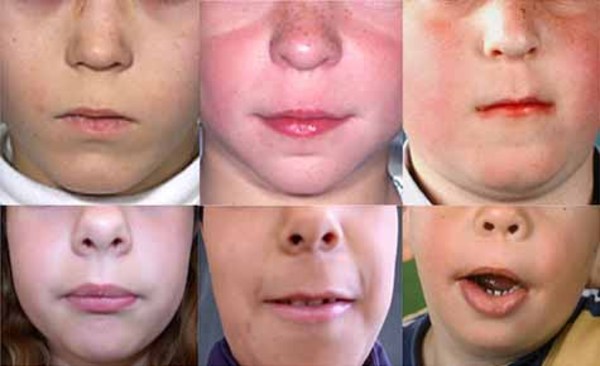
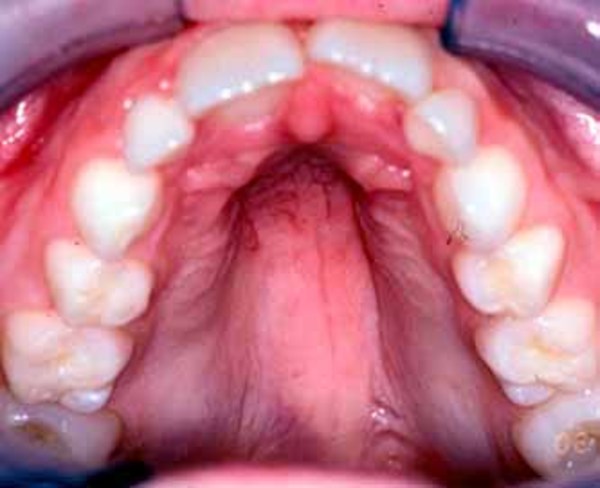
People with lateral meningocele syndrome typically have a particular pattern of facial features that may include high arched eyebrows, widely spaced eyes (hypertelorism), outside corners of the eyes that point downward (downslanting palpebral fissures), and droopy eyelids (ptosis). Affected individuals may have a flat appearance of the middle of the face and cheekbones (midface and malar hypoplasia); low-set ears; a long area between the nose and mouth (long philtrum); a thin upper lip; a high, narrow roof of the mouth, occasionally with an abnormal opening (a cleft palate); a small jaw (micrognathia); coarse hair; and a low hairline at the back of the neck.




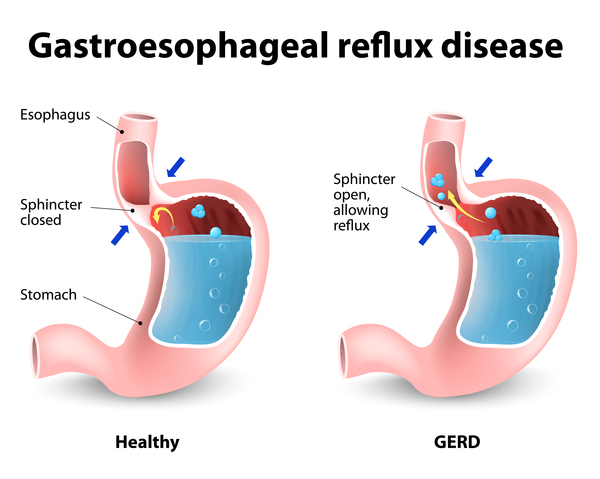
Other signs and symptoms that can occur in lateral meningocele syndrome include a high and nasal voice, hearing loss, abnormalities of the heart or the genitourinary system, poor feeding, difficulty swallowing (dysphagia), and backflow of stomach acids into the esophagus (called gastroesophageal reflux or GERD).
Frequency
Lateral meningocele syndrome is a very rare disorder. Only a small number of cases have been described in the medical literature.
Causes
Lateral meningocele syndrome is caused by mutations in the NOTCH3 gene. This gene provides instructions for making a protein with one end (the intracellular end) that remains inside the cell, a middle (transmembrane) section that spans the cell membrane, and another end (the extracellular end) that projects from the outer surface of the cell. The NOTCH3 protein is called a receptor protein because certain other proteins, called ligands, attach (bind) to the extracellular end of NOTCH3, fitting like a key into a lock. This binding causes detachment of the intracellular end of the NOTCH3 protein, called the NOTCH3 intracellular domain, or NICD. The NICD enters the cell nucleus and helps control the activity (transcription) of other genes.
The NOTCH3 gene mutations that cause lateral meningocele syndrome occur at the end of the gene in a region known as exon 33. These gene mutations result in a NOTCH3 protein with an abnormally short (truncated) NICD. The shortened protein is missing the portion that normally causes the breakdown of the NICD after it has performed its function in the cell nucleus and is no longer needed. As a result, the presence of the NICD in the cell is prolonged, and the protein continues to affect the activity of other genes. However, the result of this prolonged NICD activity and its connection to the specific features of lateral meningocele syndrome are not well understood.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new mutations in the gene and occur in people with no history of the disorder in their family. Occasionally, an affected person inherits the mutation from one affected parent.


Other Names for This Condition
- Lehman syndrome
- LMS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Alves D, Sampaio M, Figueiredo R, Leao M. Lateral meningocele syndrome: additional report and further evidence supporting a connective tissue basis. Am J Med Genet A. 2013 Jul;161A(7):1768-72. doi: 10.1002/ajmg.a.35968. Epub 2013 May 21. Citation on PubMed
- Chen CP. Syndromes, disorders and maternal risk factors associated with neural tube defects (II). Taiwan J Obstet Gynecol. 2008 Mar;47(1):10-7. doi: 10.1016/S1028-4559(08)60049-2. Citation on PubMed
- Chen KM, Bird L, Barnes P, Barth R, Hudgins L. Lateral meningocele syndrome: vertical transmission and expansion of the phenotype. Am J Med Genet A. 2005 Mar 1;133A(2):115-21. doi: 10.1002/ajmg.a.30526. Citation on PubMed
- Gripp KW, Robbins KM, Sobreira NL, Witmer PD, Bird LM, Avela K, Makitie O, Alves D, Hogue JS, Zackai EH, Doheny KF, Stabley DL, Sol-Church K. Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome. Am J Med Genet A. 2015 Feb;167A(2):271-81. doi: 10.1002/ajmg.a.36863. Epub 2014 Nov 13. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.