

Description
Laron syndrome is a rare form of short stature that results from the body's inability to use growth hormone, a substance produced by the brain's pituitary gland that helps promote growth. Affected individuals are close to normal size at birth, but they experience slow growth from early childhood that results in very short stature. If the condition is not treated, adult males typically reach a maximum height of about 4.5 feet; adult females may be just over 4 feet tall.
Other features of untreated Laron syndrome include reduced muscle strength and endurance, low blood glucose levels (hypoglycemia) in infancy, small genitals and delayed puberty, hair that is thin and fragile, and dental abnormalities. Many affected individuals have a distinctive facial appearance, including a protruding forehead, a sunken bridge of the nose (saddle nose), and a blue tint to the whites of the eyes (blue sclerae). Affected individuals have short limbs compared to the size of their torso, as well as small hands and feet. Adults with this condition tend to develop obesity. However, the signs and symptoms of Laron syndrome vary, even among affected members of the same family.
Studies suggest that people with Laron syndrome have a significantly reduced risk of cancer and type 2 diabetes. Affected individuals appear to develop these common diseases much less frequently than their unaffected relatives, despite having obesity (a risk factor for both cancer and type 2 diabetes). However, people with Laron syndrome do not seem to have an increased lifespan compared with their unaffected relatives.
Frequency
Laron syndrome is a rare disorder. About 350 people have been diagnosed with the condition worldwide. The largest single group of affected individuals (about 100 people) lives in an area of southern Ecuador.
Causes
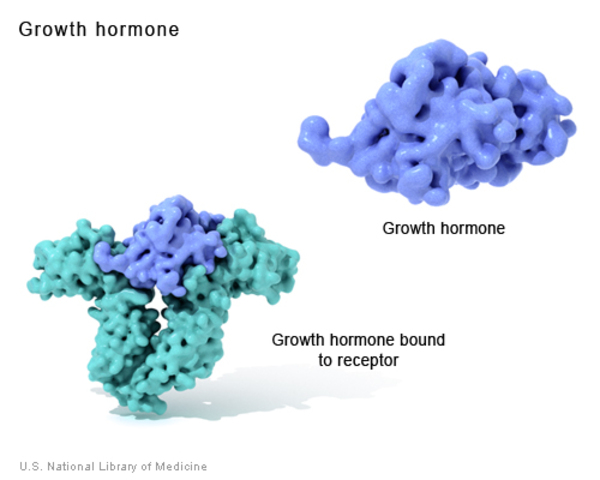
Laron syndrome is caused by mutations in the GHR gene. This gene provides instructions for making a protein called the growth hormone receptor. The receptor is present on the outer membrane of cells throughout the body, particularly liver cells. As its name suggests, the growth hormone receptor attaches (binds) to growth hormone; the two proteins fit together like a key in a lock. When growth hormone is bound to its receptor, it triggers signaling that stimulates the growth and division of cells. This signaling also leads to the production, primarily by liver cells, of another important growth-promoting hormone called insulin-like growth factor I (IGF-I).
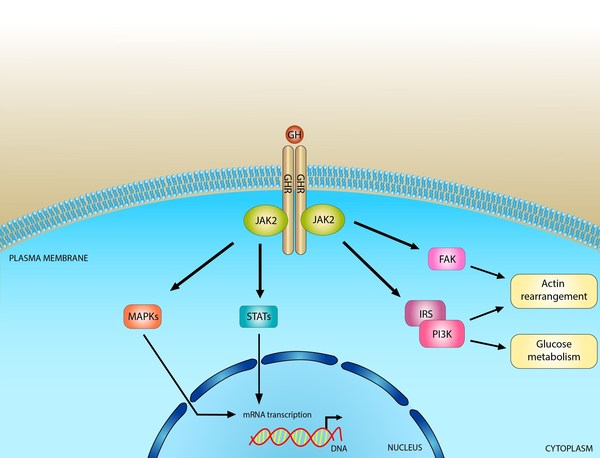
Growth hormone and IGF-I have a wide variety of effects on the growth and function of many parts of the body. For example, these hormones stimulate the growth and division of cells called chondrocytes, which play a critical role in producing new bone tissue. Growth hormone and IGF-I also influence metabolism, including how the body uses and stores carbohydrates, proteins, and fats from food.
Mutations in the GHR gene impair the receptor's ability to bind to growth hormone or to trigger signaling within cells. As a result, even when growth hormone is available, cells are unable to respond by producing IGF-I and stimulating growth and division. The cells' inability to react to growth hormone, which is described as growth hormone insensitivity, disrupts the normal growth and function of many different tissues. Short stature results when growth hormone cannot adequately stimulate the growth of bones. Changes in metabolism caused by insensitivity to growth hormone and the resulting shortage of IGF-I cause many of the other features of the condition, including obesity.
Researchers are working to determine how mutations in the GHR gene may protect people with Laron syndrome from developing cancer and type 2 diabetes. Studies suggest that insensitivity to growth hormone may help prevent the uncontrolled growth and division of cells that can lead to the development of cancerous tumors. Growth hormone insensitivity also appears to alter how the body responds to insulin, which is a hormone that regulates blood glucose levels. Resistance to the effects of insulin is a major risk factor for type 2 diabetes. People with Laron syndrome have the opposite situation, an increased sensitivity to insulin, which likely helps explain their reduced risk of this common disease.
Inheritance
Most cases of Laron syndrome are inherited in an autosomal recessive pattern, which means both copies of the GHR gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Much less commonly, the condition has an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most of these cases, an affected person has one parent with the condition.

Other Names for This Condition
- GH-R deficiency
- Growth hormone insensitivity syndrome
- Growth hormone receptor defect
- Growth hormone receptor deficiency
- Laron dwarfism
- Laron-type dwarfism
- Laron-type isolated somatotropin defect
- Laron-type pituitary dwarfism
- Laron-type short stature
- Pituitary dwarfism II
- Primary GH resistance
- Primary growth hormone resistance
- Severe GH insensitivity
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Amselem S, Duquesnoy P, Attree O, Novelli G, Bousnina S, Postel-Vinay MC, Goossens M. Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med. 1989 Oct 12;321(15):989-95. doi: 10.1056/NEJM198910123211501. Citation on PubMed
- Burren CP, Woods KA, Rose SJ, Tauber M, Price DA, Heinrich U, Gilli G, Razzaghy-Azar M, Al-Ashwal A, Crock PA, Rochiccioli P, Yordam N, Ranke MB, Chatelain PG, Preece MA, Rosenfeld RG, Savage MO. Clinical and endocrine characteristics in atypical and classical growth hormone insensitivity syndrome. Horm Res. 2001;55(3):125-30. doi: 10.1159/000049983. Citation on PubMed
- David A, Hwa V, Metherell LA, Netchine I, Camacho-Hubner C, Clark AJ, Rosenfeld RG, Savage MO. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. 2011 Aug;32(4):472-97. doi: 10.1210/er.2010-0023. Epub 2011 Apr 27. Citation on PubMed
- Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, Hwang D, Martin-Montalvo A, Saavedra J, Ingles S, de Cabo R, Cohen P, Longo VD. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011 Feb 16;3(70):70ra13. doi: 10.1126/scitranslmed.3001845. Citation on PubMed or Free article on PubMed Central
- Guevara-Aguirre J, Rosenbloom AL. Obesity, diabetes and cancer: insight into the relationship from a cohort with growth hormone receptor deficiency. Diabetologia. 2015 Jan;58(1):37-42. doi: 10.1007/s00125-014-3397-3. Epub 2014 Oct 15. Citation on PubMed
- Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003. J Clin Endocrinol Metab. 2004 Mar;89(3):1031-44. doi: 10.1210/jc.2003-031033. Citation on PubMed
- Shevah O, Laron Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: a preliminary report. Growth Horm IGF Res. 2007 Feb;17(1):54-7. doi: 10.1016/j.ghir.2006.10.007. Epub 2006 Dec 12. Citation on PubMed
- Wit JM, Oostdijk W, Losekoot M. Spectrum of insulin-like growth factor deficiency. Endocr Dev. 2012;23:30-41. doi: 10.1159/000341739. Epub 2012 Nov 23. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.