

Description
Lafora progressive myoclonus epilepsy is a brain disorder characterized by recurrent seizures (epilepsy) and a decline in intellectual function. The signs and symptoms of the disorder usually appear in late childhood or adolescence and worsen with time.
Myoclonus is a term used to describe episodes of sudden, involuntary muscle jerking or twitching that can affect part of the body or the entire body. Myoclonus can occur when an affected person is at rest, and it is made worse by motion, excitement, or flashing light (photic stimulation). In the later stages of Lafora progressive myoclonus epilepsy, myoclonus often occurs continuously and affects the entire body.
Several types of seizures commonly occur in people with Lafora progressive myoclonus epilepsy. Generalized tonic-clonic seizures (also known as grand mal seizures) affect the entire body, causing muscle rigidity, convulsions, and loss of consciousness. Affected individuals may also experience occipital seizures, which can cause temporary blindness and visual hallucinations. Over time, the seizures worsen and become more difficult to treat. A life-threatening seizure condition called status epilepticus may also develop. Status epilepticus is a continuous state of seizure activity lasting longer than several minutes.
About the same time seizures begin, intellectual function starts to decline. Behavioral changes, depression, confusion, and speech difficulties (dysarthria) are among the early signs and symptoms of this disorder. As the condition worsens, a continued loss of intellectual function (dementia) impairs memory, judgment, and thought. Affected people lose the ability to perform the activities of daily living by their mid-twenties, and they ultimately require comprehensive care. People with Lafora progressive myoclonus epilepsy generally survive up to 10 years after symptoms first appear.
Frequency
The prevalence of Lafora progressive myoclonus epilepsy is unknown. Although the condition occurs worldwide, it appears to be most common in Mediterranean countries (including Spain, France, and Italy), parts of Central Asia, India, Pakistan, North Africa, and the Middle East.
Causes
Lafora progressive myoclonus epilepsy can be caused by mutations in either the EPM2A gene or the NHLRC1 gene. These genes provide instructions for making proteins called laforin and malin, respectively. Laforin and malin play a critical role in the survival of nerve cells (neurons) in the brain.

Studies suggest that laforin and malin work together and may have several functions. One of these is to help regulate the production of a complex sugar called glycogen, which is a major source of stored energy in the body. The body stores this sugar in the liver and muscles, breaking it down when it is needed for fuel. Laforin and malin may prevent a potentially damaging buildup of glycogen in tissues that do not normally store this molecule, such as those of the nervous system.
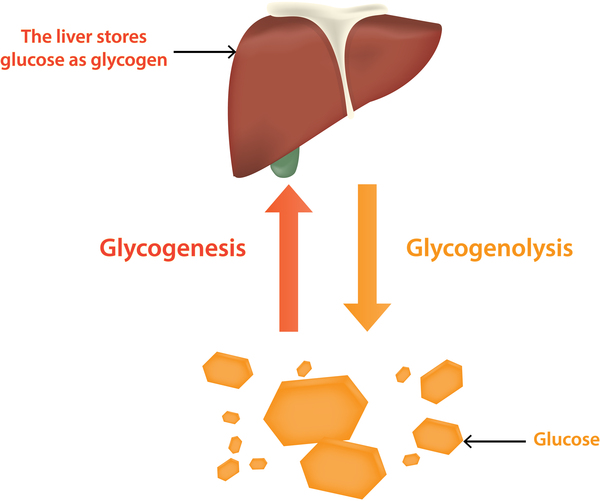
Researchers have discovered that people with Lafora progressive myoclonus epilepsy have distinctive clumps called Lafora bodies within their cells. Lafora bodies are made up of an abnormal form of glycogen that cannot be broken down and used for fuel. Instead, it builds up to form clumps that can damage cells. Neurons appear to be particularly vulnerable to this type of damage. Although Lafora bodies are found in many of the body's tissues, the signs and symptoms of Lafora progressive myoclonus epilepsy are limited to the nervous system.
Mutations in the EPM2A gene prevent cells from making functional laforin, while NHLRC1 gene mutations prevent the production of functional malin. It is unclear how a loss of either of these proteins leads to the formation of Lafora bodies. However, a loss of laforin or malin ultimately results in the death of neurons, which interferes with the brain's normal functions. The condition tends to progress more slowly in some people with NHLRC1 gene mutations than in those with EPM2A gene mutations.
Mutations in the EPM2A and NHLRC1 genes account for 80 percent to 90 percent of all cases of Lafora progressive myoclonus epilepsy. In the remaining cases, the cause of the condition is unknown. Researchers are searching for other genetic changes that may underlie this disease.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Epilepsy, progressive myoclonic, Lafora
- Lafora body disease
- Lafora disease
- Lafora progressive myoclonic epilepsy
- Lafora type progressive myoclonic epilepsy
- Myoclonic epilepsy of Lafora
- Progressive myoclonic epilepsy type 2
- Progressive myoclonus epilepsy, Lafora type
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Andrade DM, Turnbull J, Minassian BA. Lafora disease, seizures and sugars. Acta Myol. 2007 Jul;26(1):83-6. Citation on PubMed or Free article on PubMed Central
- Delgado-Escueta AV. Advances in lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep. 2007 Sep;7(5):428-33. doi: 10.1007/s11910-007-0066-7. Citation on PubMed
- Ganesh S, Puri R, Singh S, Mittal S, Dubey D. Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J Hum Genet. 2006;51(1):1-8. doi: 10.1007/s10038-005-0321-1. Epub 2005 Nov 26. Citation on PubMed
- Girard JM, Turnbull J, Ramachandran N, Minassian BA. Progressive myoclonus epilepsy. Handb Clin Neurol. 2013;113:1731-6. doi: 10.1016/B978-0-444-59565-2.00043-5. Citation on PubMed
- Jansen AC, Andermann E. Progressive Myoclonus Epilepsy, Lafora Type. 2007 Dec 28 [updated 2019 Feb 21]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1389/ Citation on PubMed
- Kalviainen R. Progressive Myoclonus Epilepsies. Semin Neurol. 2015 Jun;35(3):293-9. doi: 10.1055/s-0035-1552620. Epub 2015 Jun 10. Citation on PubMed
- Lohi H, Turnbull J, Zhao XC, Pullenayegum S, Ianzano L, Yahyaoui M, Mikati MA, Quinn NP, Franceschetti S, Zara F, Minassian BA. Genetic diagnosis in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology. 2007 Mar 27;68(13):996-1001. doi: 10.1212/01.wnl.0000258561.02248.2f. Erratum In: Neurology. 2007 Jun 12;68(24):2153. Quinn, NP [added]. Citation on PubMed
- Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum Mutat. 2009 May;30(5):715-23. doi: 10.1002/humu.20954. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.