

Description
Koolen-de Vries syndrome is a disorder characterized by developmental delay and mild to moderate intellectual disability. People with this disorder typically have a disposition that is described as cheerful, sociable, and cooperative. They usually have weak muscle tone (hypotonia) in childhood. About half have recurrent seizures (epilepsy).
Affected individuals often have distinctive facial features including a high, broad forehead; droopy eyelids (ptosis); a narrowing of the eye openings (blepharophimosis); outer corners of the eyes that point upward (upward-slanting palpebral fissures); skin folds covering the inner corner of the eyes (epicanthal folds); a bulbous nose; and prominent ears. Males with Koolen-de Vries syndrome often have undescended testes (cryptorchidism). Defects in the walls between the chambers of the heart (septal defects) or other cardiac abnormalities, kidney problems, and skeletal anomalies such as foot deformities occur in some affected individuals.
Frequency
The prevalence of Koolen-de Vries syndrome is estimated to be 1 in 16,000. However, the underlying genetic cause is often not identified in people with intellectual disability, so this condition is likely underdiagnosed.
Causes
Koolen-de Vries syndrome is caused by genetic changes that eliminate the function of one copy of the KANSL1 gene in each cell. Most affected individuals are missing a small amount of genetic material, including the KANSL1 gene, from one copy of chromosome 17. This type of genetic abnormality is called a microdeletion. A small number of individuals with Koolen-de Vries syndrome do not have a chromosome 17 microdeletion but instead have a mutation within the KANSL1 gene that causes one copy of the gene to be nonfunctional.
The microdeletion that causes Koolen-de Vries syndrome occurs on the long (q) arm of chromosome 17 at a location designated q21.31. While the exact size of the deletion varies among affected individuals, most are missing a sequence of about 500,000 DNA building blocks (base pairs) containing several genes. However, because individuals with KANSL1 gene mutations have the same signs and symptoms as those with the microdeletion, researchers have concluded that the loss of this gene accounts for the features of this disorder.
The KANSL1 gene provides instructions for making a protein that helps regulate gene activity (expression) by modifying chromatin. Chromatin is the complex of DNA and protein that packages DNA into chromosomes. The protein produced from the KANSL1 gene is found in most organs and tissues of the body before birth and throughout life. By its involvement in controlling the activity of other genes, this protein plays an important role in the development and function of many parts of the body. Loss of one copy of this gene impairs normal development and function, but the relationship of KANSL1 gene loss to the specific signs and symptoms of Koolen-de Vries syndrome is unclear.
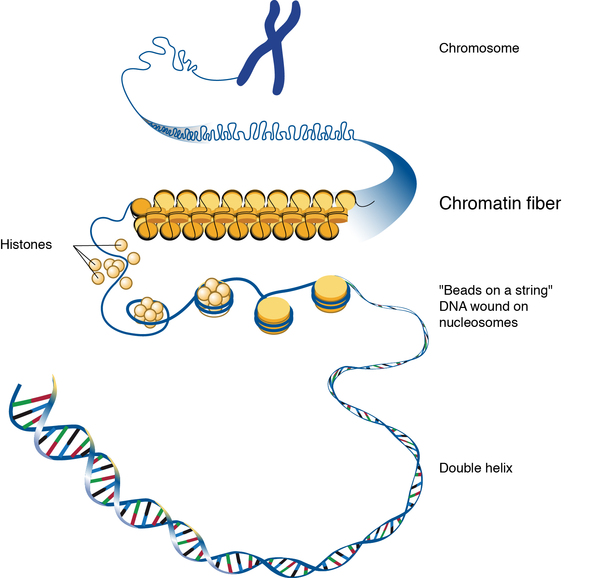
Inheritance
Koolen-de Vries syndrome is considered an autosomal dominant condition because a deletion or mutation affecting one copy of the KANSL1 gene in each cell is sufficient to cause the disorder. In most cases, the disorder is not inherited. The genetic change occurs most often as a random event during the formation of reproductive cells (eggs and sperm) or in early fetal development. Affected people typically have no history of the disorder in their family. While it is possible for them to pass the condition on to their children, no individuals with Koolen-de Vries syndrome have been known to reproduce.

Most people with Koolen-de Vries syndrome caused by a deletion have had at least one parent with a common variant of the 17q21.31 region of chromosome 17 called the H2 lineage. This variant is found in 20 percent of people of European and Middle Eastern descent, although it is rare in other populations. In the H2 lineage, a 900 kb segment of DNA, which includes the region deleted in most cases of Koolen-de Vries syndrome, has undergone an inversion. An inversion involves two breaks in a chromosome; the resulting piece of DNA is reversed and reinserted into the chromosome.
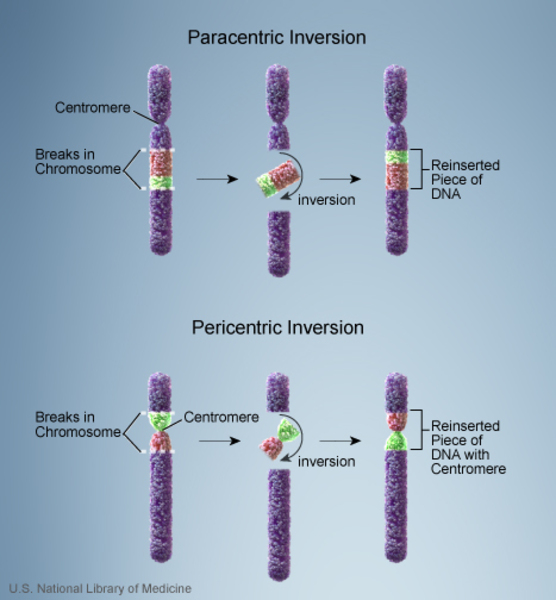
People with the H2 lineage have no health problems related to the inversion. However, genetic material can be lost or duplicated when the inversion is passed to the next generation. Other, unknown factors are thought to play a role in this process. So while the inversion is very common, only an extremely small percentage of parents with the inversion have a child affected by Koolen-de Vries syndrome.
Other Names for This Condition
- 17q21.31 deletion syndrome
- 17q21.31 microdeletion syndrome
- Chromosome 17q21.31 microdeletion syndrome
- KANSL1-related intellectual disability syndrome
- KDVS
- Koolen syndrome
- Microdeletion 17q21.31 syndrome
- Monosomy 17q21.31
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dubourg C, Sanlaville D, Doco-Fenzy M, Le Caignec C, Missirian C, Jaillard S, Schluth-Bolard C, Landais E, Boute O, Philip N, Toutain A, David A, Edery P, Moncla A, Martin-Coignard D, Vincent-Delorme C, Mortemousque I, Duban-Bedu B, Drunat S, Beri M, Mosser J, Odent S, David V, Andrieux J. Clinical and molecular characterization of 17q21.31 microdeletion syndrome in 14 French patients with mental retardation. Eur J Med Genet. 2011 Mar-Apr;54(2):144-51. doi: 10.1016/j.ejmg.2010.11.003. Epub 2010 Nov 20. Citation on PubMed
- Egger JI, Wingbermuhle E, Verhoeven WM, Dijkman M, Radke S, de Bruijn ER, de Vries B, Kessels RP, Koolen D. Hypersociability in the behavioral phenotype of 17q21.31 microdeletion syndrome. Am J Med Genet A. 2013 Jan;161A(1):21-6. doi: 10.1002/ajmg.a.35652. Epub 2012 Nov 20. Citation on PubMed
- Itsara A, Vissers LE, Steinberg KM, Meyer KJ, Zody MC, Koolen DA, de Ligt J, Cuppen E, Baker C, Lee C, Graves TA, Wilson RK, Jenkins RB, Veltman JA, Eichler EE. Resolving the breakpoints of the 17q21.31 microdeletion syndrome with next-generation sequencing. Am J Hum Genet. 2012 Apr 6;90(4):599-613. doi: 10.1016/j.ajhg.2012.02.013. Citation on PubMed or Free article on PubMed Central
- Koolen DA, Kramer JM, Neveling K, Nillesen WM, Moore-Barton HL, Elmslie FV, Toutain A, Amiel J, Malan V, Tsai AC, Cheung SW, Gilissen C, Verwiel ET, Martens S, Feuth T, Bongers EM, de Vries P, Scheffer H, Vissers LE, de Brouwer AP, Brunner HG, Veltman JA, Schenck A, Yntema HG, de Vries BB. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat Genet. 2012 Apr 29;44(6):639-41. doi: 10.1038/ng.2262. Citation on PubMed
- Koolen DA, Morgan A, de Vries BBA. Koolen-de Vries Syndrome. 2010 Jan 26 [updated 2023 Feb 2]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK24676/ Citation on PubMed
- Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, Saugier-Veber P, Pfundt R, Vissers LE, Destree A, Grisart B, Rooms L, Van der Aa N, Field M, Hackett A, Bell K, Nowaczyk MJ, Mancini GM, Poddighe PJ, Schwartz CE, Rossi E, De Gregori M, Antonacci-Fulton LL, McLellan MD 2nd, Garrett JM, Wiechert MA, Miner TL, Crosby S, Ciccone R, Willatt L, Rauch A, Zenker M, Aradhya S, Manning MA, Strom TM, Wagenstaller J, Krepischi-Santos AC, Vianna-Morgante AM, Rosenberg C, Price SM, Stewart H, Shaw-Smith C, Brunner HG, Wilkie AO, Veltman JA, Zuffardi O, Eichler EE, de Vries BB. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008 Nov;45(11):710-20. doi: 10.1136/jmg.2008.058701. Epub 2008 Jul 15. Erratum In: J Med Genet. 2009 Aug;46(8):576. Citation on PubMed or Free article on PubMed Central
- Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid BM, Schoumans J, Knoers NV, van Kessel AG, Sistermans EA, Veltman JA, Brunner HG, de Vries BB. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006 Sep;38(9):999-1001. doi: 10.1038/ng1853. Epub 2006 Aug 13. Citation on PubMed
- Sharkey FH, Morrison N, Murray R, Iremonger J, Stephen J, Maher E, Tolmie J, Jackson AP. 17q21.31 microdeletion syndrome: further expanding the clinical phenotype. Cytogenet Genome Res. 2009;127(1):61-6. doi: 10.1159/000279260. Epub 2010 Jan 27. Citation on PubMed
- Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, Desnica N, Hicks A, Gylfason A, Gudbjartsson DF, Jonsdottir GM, Sainz J, Agnarsson K, Birgisdottir B, Ghosh S, Olafsdottir A, Cazier JB, Kristjansson K, Frigge ML, Thorgeirsson TE, Gulcher JR, Kong A, Stefansson K. A common inversion under selection in Europeans. Nat Genet. 2005 Feb;37(2):129-37. doi: 10.1038/ng1508. Epub 2005 Jan 16. Citation on PubMed
- Tan TY, Aftimos S, Worgan L, Susman R, Wilson M, Ghedia S, Kirk EP, Love D, Ronan A, Darmanian A, Slavotinek A, Hogue J, Moeschler JB, Ozmore J, Widmer R, Bruno D, Savarirayan R, Peters G. Phenotypic expansion and further characterisation of the 17q21.31 microdeletion syndrome. J Med Genet. 2009 Jul;46(7):480-9. doi: 10.1136/jmg.2008.065391. Epub 2009 May 15. Erratum In: J Med Genet. 2009 Aug;46(8):576. Bruno, Damien [added]. Citation on PubMed
- Zollino M, Orteschi D, Murdolo M, Lattante S, Battaglia D, Stefanini C, Mercuri E, Chiurazzi P, Neri G, Marangi G. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat Genet. 2012 Apr 29;44(6):636-8. doi: 10.1038/ng.2257. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.