

Description
Klippel-Feil syndrome is a bone disorder characterized by the abnormal joining (fusion) of two or more spinal bones in the neck (cervical vertebrae). The vertebral fusion is present from birth. Three major features result from this vertebral fusion: a short neck, the resulting appearance of a low hairline at the back of the head, and a limited range of motion in the neck. Most affected people have one or two of these characteristic features. Less than half of all individuals with Klippel-Feil syndrome have all three classic features of this condition.
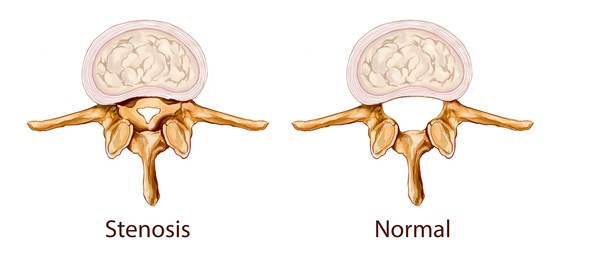
In people with Klippel-Feil syndrome, the fused vertebrae can limit the range of movement of the neck and back as well as lead to chronic headaches and muscle pain in the neck and back that range in severity. People with minimal bone involvement often have fewer problems compared to individuals with several vertebrae affected. The shortened neck can cause a slight difference in the size and shape of the right and left sides of the face (facial asymmetry). Trauma to the spine, such as a fall or car accident, can aggravate problems in the fused area. Fusion of the vertebrae can lead to nerve damage in the head, neck, or back. Over time, individuals with Klippel-Feil syndrome can develop a narrowing of the spinal canal (spinal stenosis) in the neck, which can compress and damage the spinal cord. Rarely, spinal nerve abnormalities may cause abnormal sensations or involuntary movements in people with Klippel-Feil syndrome. Affected individuals may develop a painful joint disorder called osteoarthritis around the areas of fused bone or experience painful involuntary tensing of the neck muscles (cervical dystonia). In addition to the fused cervical bones, people with this condition may have abnormalities in other vertebrae. Many people with Klippel-Feil syndrome have abnormal side-to-side curvature of the spine (scoliosis) due to malformation of the vertebrae; fusion of additional vertebrae below the neck may also occur.

People with Klippel-Feil syndrome may have a wide variety of other features in addition to their spine abnormalities. Some people with this condition have hearing difficulties, eye abnormalities, an opening in the roof of the mouth (cleft palate), genitourinary problems such as abnormal kidneys or reproductive organs, heart abnormalities, or lung defects that can cause breathing problems. Affected individuals may have other skeletal defects including arms or legs of unequal length (limb length discrepancy), which can result in misalignment of the hips or knees. Additionally, the shoulder blades may be underdeveloped so that they sit abnormally high on the back, a condition called Sprengel deformity. Rarely, structural brain abnormalities or a type of birth defect that occurs during the development of the brain and spinal cord (neural tube defect) can occur in people with Klippel-Feil syndrome.

In some cases, Klippel-Feil syndrome occurs as a feature of another disorder or syndrome, such as Wildervanck syndrome or hemifacial microsomia. In these instances, affected individuals have the signs and symptoms of both Klippel-Feil syndrome and the additional disorder.
Frequency
Klippel-Feil syndrome is estimated to occur in 1 in 40,000 to 42,000 newborns worldwide. Females seem to be affected slightly more often than males.
Causes
Mutations in the GDF6, GDF3, or MEOX1 gene can cause Klippel-Feil syndrome. These genes are involved in proper bone development. The protein produced from the GDF6 gene is necessary for the formation of bones and joints, including those in the spine. While the protein produced from the GDF3 gene is known to be involved in bone development, its exact role is unclear. The protein produced from the MEOX1 gene, called homeobox protein MOX-1, regulates the process that begins separating vertebrae from one another during early development.
GDF6 and GDF3 gene mutations that cause Klippel-Feil syndrome likely lead to reduced function of the respective proteins. MEOX1 gene mutations lead to a complete lack of homeobox protein MOX-1. Although the GDF6, GDF3, and homeobox protein MOX-1 proteins are involved in bone development, particularly formation of vertebrae, it is unclear how a shortage of one of these proteins leads to incomplete separation of the cervical vertebrae in people with Klippel-Feil syndrome.
When Klippel-Feil syndrome is a feature of another disorder, it is caused by mutations in genes involved in the other disorder.
Inheritance
When Klippel-Feil syndrome is caused by mutations in the GDF6 or GDF3 genes, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

When caused by mutations in the MEOX1 gene, Klippel-Feil syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

As a feature of another disorder, Klippel-Feil syndrome is inherited in whatever pattern the other disorder follows.
Other Names for This Condition
- Cervical fusion syndrome
- Cervical vertebral fusion
- Cervical vertebral fusion syndrome
- Congenital dystrophia brevicollis
- Dystrophia brevicollis congenita
- Fusion of cervical vertebrae
- KFS
- Klippel-Feil deformity
- Klippel-Feil sequence
- Vertebral cervical fusion syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bayrakli F, Guclu B, Yakicier C, Balaban H, Kartal U, Erguner B, Sagiroglu MS, Yuksel S, Ozturk AR, Kazanci B, Ozum U, Kars HZ. Mutation in MEOX1 gene causes a recessive Klippel-Feil syndrome subtype. BMC Genet. 2013 Sep 28;14:95. doi: 10.1186/1471-2156-14-95. Citation on PubMed or Free article on PubMed Central
- Klimo P Jr, Rao G, Brockmeyer D. Congenital anomalies of the cervical spine. Neurosurg Clin N Am. 2007 Jul;18(3):463-78. doi: 10.1016/j.nec.2007.04.005. Citation on PubMed
- Lampropoulou-Adamidou K, Athanassacopoulos M, Karampinas PK, Vlamis J, Korres DS, Pneumaticos SG. Congenital variations of the upper cervical spine and their importance in preoperative diagnosis. A case report and a review of the literature. Eur J Orthop Surg Traumatol. 2013 Jul;23 Suppl 1:S101-5. doi: 10.1007/s00590-013-1216-z. Epub 2013 Apr 6. Citation on PubMed
- Mohamed JY, Faqeih E, Alsiddiky A, Alshammari MJ, Ibrahim NA, Alkuraya FS. Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. Am J Hum Genet. 2013 Jan 10;92(1):157-61. doi: 10.1016/j.ajhg.2012.11.016. Epub 2013 Jan 3. Citation on PubMed or Free article on PubMed Central
- Samartzis D, Kalluri P, Herman J, Lubicky JP, Shen FH. The extent of fusion within the congenital Klippel-Feil segment. Spine (Phila Pa 1976). 2008 Jul 1;33(15):1637-42. doi: 10.1097/BRS.0b013e31817c0bc2. Citation on PubMed
- Samartzis D, Lubicky JP, Herman J, Shen FH. Faces of Spine Care: From the Clinic and Imaging Suite. Klippel-Feil syndrome and associated abnormalities: the necessity for a multidisciplinary approach in patient management. Spine J. 2007 Jan-Feb;7(1):135-7. doi: 10.1016/j.spinee.2006.05.019. No abstract available. Citation on PubMed
- Thomsen M, Krober M, Schneider U, Carstens C. Congenital limb deficiences associated with Klippel-Feil syndrome: a survey of 57 subjects. Acta Orthop Scand. 2000 Oct;71(5):461-4. doi: 10.1080/000164700317381135. Citation on PubMed
- Tracy MR, Dormans JP, Kusumi K. Klippel-Feil syndrome: clinical features and current understanding of etiology. Clin Orthop Relat Res. 2004 Jul;(424):183-90. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.