

Description
KBG syndrome is a rare disorder that affects several body systems. "KBG" represents the surname initials of the first families diagnosed with the disorder. Common signs and symptoms in individuals with this condition include unusual facial features, skeletal abnormalities, and intellectual disability.
A characteristic feature of KBG syndrome is unusually large upper front teeth (macrodontia). Other distinctive facial features include a wide, short skull (brachycephaly), a triangular face shape, widely spaced eyes (hypertelorism), wide eyebrows that may grow together in the middle (synophrys), a prominent nasal bridge, a long space between the nose and upper lip (long philtrum), and a thin upper lip.
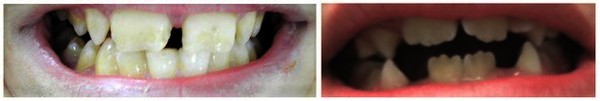

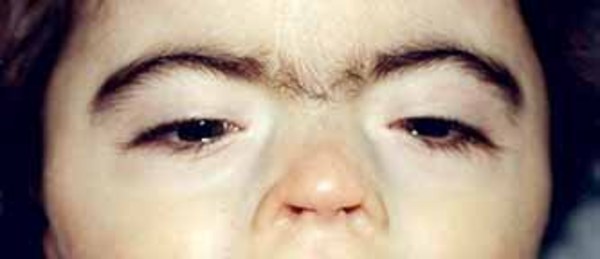
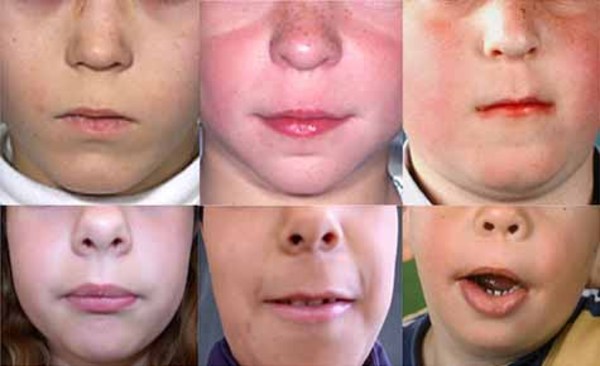

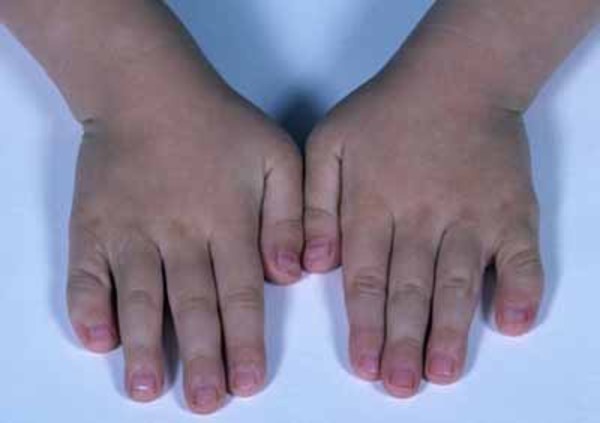
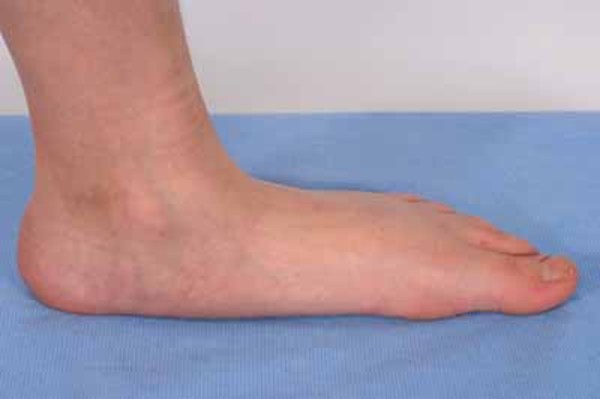
A common skeletal abnormality in people with KBG syndrome is slowed mineralization of bones (delayed bone age); for example, an affected 3-year-old child may have bones more typical of a child of 2. In addition, affected individuals can have abnormalities of the bones of the spine (vertebrae) and ribs. They can also have abnormalities of the bones of the hands or feet, including unusually short or curved fifth (pinky) fingers (brachydactyly or clinodactyly, respectively) and flat feet (pes planus). Most affected individuals are shorter than average from birth.

Development of mental and movement abilities is also delayed in KBG syndrome. Most affected individuals learn to speak and walk later than normal and have mild to moderate intellectual disability. Most people with this condition have neurodevelopmental disorders, such as hyperactivity; anxiety; or autism spectrum disorder, which is characterized by impaired communication and social interactions.
Less common features of KBG syndrome include hearing loss, seizures, and heart defects.
Frequency
KBG syndrome is a rare disorder that has been reported in more than 150 individuals in the medical literature, though there are likely more who have not been recorded in the literature. For unknown reasons, males are affected more often than females. Doctors think the disorder is underdiagnosed because the signs and symptoms can be mild and may be attributed to other disorders.
Causes
KBG syndrome is caused by mutations in the ANKRD11 gene. The protein produced from this gene enables other proteins to interact with each other and helps control gene activity. The ANKRD11 protein is found in nerve cells (neurons) in the brain. It plays a role in the proper development of the brain and may be involved in the ability of neurons to change and adapt over time (plasticity), which is important for learning and memory. ANKRD11 may function in other cells in the body and appears to be involved in normal bone development.
Most of the ANKRD11 gene mutations involved in KBG syndrome lead to an abnormally short ANKRD11 protein, which likely has little or no function. Reduction of this protein's function is thought to underlie the signs and symptoms of the condition. Because ANKRD11 is thought to play an important role in neurons and brain development, researchers speculate that a partial loss of its function may lead to developmental delay and intellectual disability in KBG syndrome. However, the mechanism is not fully known. It is also unclear how loss of ANKRD11 function leads to the skeletal features of the condition.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Other Names for This Condition
- Macrodontia, mental retardation, characteristic facies, short stature, and skeletal anomalies
- Short stature, characteristic facies, macrodontia, mental retardation, and skeletal anomalies
- Short stature-characteristic facies-mental retardation-macrodontia-skeletal anomalies syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Gallagher D, Voronova A, Zander MA, Cancino GI, Bramall A, Krause MP, Abad C, Tekin M, Neilsen PM, Callen DF, Scherer SW, Keller GM, Kaplan DR, Walz K, Miller FD. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev Cell. 2015 Jan 12;32(1):31-42. doi: 10.1016/j.devcel.2014.11.031. Epub 2014 Dec 31. Citation on PubMed
- Kim HJ, Cho E, Park JB, Im WY, Kim HJ. A Korean family with KBG syndrome identified by ANKRD11 mutation, and phenotypic comparison of ANKRD11 mutation and 16q24.3 microdeletion. Eur J Med Genet. 2015 Feb;58(2):86-94. doi: 10.1016/j.ejmg.2014.11.003. Epub 2014 Nov 20. Citation on PubMed
- Kleyner R, Malcolmson J, Tegay D, Ward K, Maughan A, Maughan G, Nelson L, Wang K, Robison R, Lyon GJ. KBG syndrome involving a single-nucleotide duplication in ANKRD11. Cold Spring Harb Mol Case Stud. 2016 Nov;2(6):a001131. doi: 10.1101/mcs.a001131. Citation on PubMed or Free article on PubMed Central
- Lo-Castro A, Brancati F, Digilio MC, Garaci FG, Bollero P, Alfieri P, Curatolo P. Neurobehavioral phenotype observed in KBG syndrome caused by ANKRD11 mutations. Am J Med Genet B Neuropsychiatr Genet. 2013 Jan;162B(1):17-23. doi: 10.1002/ajmg.b.32113. Epub 2012 Nov 26. Citation on PubMed
- Morel Swols D, Foster J 2nd, Tekin M. KBG syndrome. Orphanet J Rare Dis. 2017 Dec 19;12(1):183. doi: 10.1186/s13023-017-0736-8. Citation on PubMed or Free article on PubMed Central
- Ockeloen CW, Willemsen MH, de Munnik S, van Bon BW, de Leeuw N, Verrips A, Kant SG, Jones EA, Brunner HG, van Loon RL, Smeets EE, van Haelst MM, van Haaften G, Nordgren A, Malmgren H, Grigelioniene G, Vermeer S, Louro P, Ramos L, Maal TJ, van Heumen CC, Yntema HG, Carels CE, Kleefstra T. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur J Hum Genet. 2015 Sep;23(9):1176-85. doi: 10.1038/ejhg.2014.253. Epub 2014 Nov 26. Erratum In: Eur J Hum Genet. 2015 Sep;23(9):1270. Citation on PubMed or Free article on PubMed Central
- Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, Bademci G, Agolini E, Guo S, Konuk B, Kavaz A, Blanton S, Digilio MC, Dallapiccola B, Young J, Zuchner S, Tekin M. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011 Aug 12;89(2):289-94. doi: 10.1016/j.ajhg.2011.06.007. Epub 2011 Jul 21. Citation on PubMed or Free article on PubMed Central
- Walz K, Cohen D, Neilsen PM, Foster J 2nd, Brancati F, Demir K, Fisher R, Moffat M, Verbeek NE, Bjorgo K, Lo Castro A, Curatolo P, Novelli G, Abad C, Lei C, Zhang L, Diaz-Horta O, Young JI, Callen DF, Tekin M. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum Genet. 2015 Feb;134(2):181-90. doi: 10.1007/s00439-014-1509-2. Epub 2014 Nov 21. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.