

Description
Idiopathic pulmonary fibrosis is a chronic, progressive lung disease. This condition causes scar tissue (fibrosis) to build up in the lungs, which makes the lungs unable to transport oxygen into the bloodstream effectively. The disease usually affects people between the ages of 50 and 70. Idiopathic pulmonary fibrosis belongs to a group of conditions called interstitial lung diseases (also known as ILD), which describes lung diseases that involve inflammation or scarring in the lung.
The most common signs and symptoms of idiopathic pulmonary fibrosis are shortness of breath and a persistent dry, hacking cough. Many affected individuals also experience a loss of appetite and gradual weight loss. Some people with idiopathic pulmonary fibrosis develop widened and rounded tips of the fingers and toes (clubbing) resulting from a shortage of oxygen. These features are relatively nonspecific; not everyone with these health problems has idiopathic pulmonary fibrosis. Other respiratory diseases, some of which are less serious, can cause similar signs and symptoms.
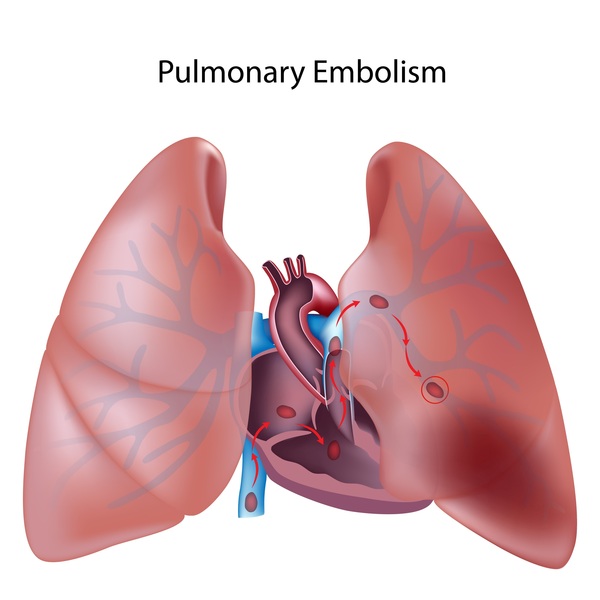
In people with idiopathic pulmonary fibrosis, scarring of the lungs increases over time until the lungs can no longer provide enough oxygen to the body's organs and tissues. Some people with idiopathic pulmonary fibrosis develop other serious lung conditions, including lung cancer, blood clots in the lungs (pulmonary emboli), pneumonia, or high blood pressure in the blood vessels that supply the lungs (pulmonary hypertension). Most affected individuals survive 3 to 5 years after their diagnosis. However, the course of the disease is highly variable; some affected people become seriously ill within a few months, while others may live with the disease for a decade or longer.
In most cases, idiopathic pulmonary fibrosis occurs in only one person in a family. These cases are described as sporadic. However, a small percentage of people with this disease have at least one other affected family member. When idiopathic pulmonary fibrosis occurs in multiple members of the same family, it is known as familial pulmonary fibrosis.
Frequency
Idiopathic pulmonary fibrosis has an estimated prevalence of 13 to 20 per 100,000 people worldwide. About 100,000 people are affected in the United States, and 30,000 to 40,000 new cases are diagnosed each year.
Familial pulmonary fibrosis is less common than the sporadic form of the disease. Only a small percentage of cases of idiopathic pulmonary fibrosis appear to run in families.
Causes
The cause of idiopathic pulmonary fibrosis is unknown. The fibrosis that builds up in the lungs is thought to develop as a result of abnormal tissue repair following tissue damage. This abnormal repair response probably results from a combination of genetic and environmental factors. It is likely that genetic changes increase a person's risk of developing idiopathic pulmonary fibrosis, and then exposure to certain environmental factors triggers the disease.
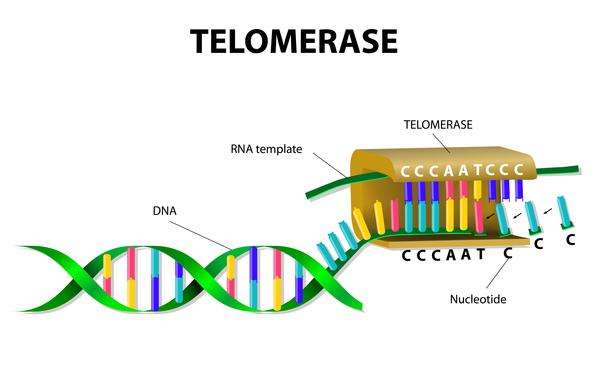
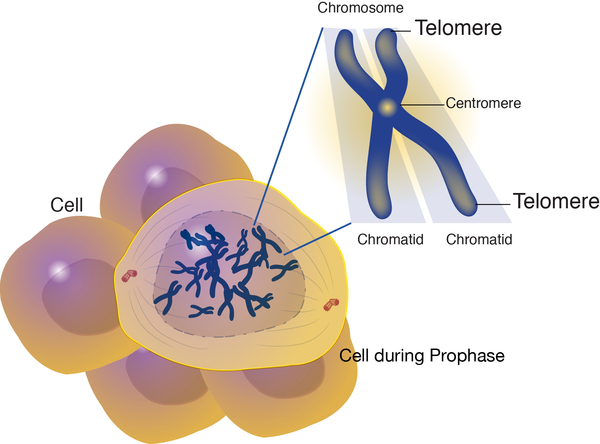
Changes in several genes have been suggested as risk factors for idiopathic pulmonary fibrosis, while it is likely that other genetic influences have yet to be discovered. Most of the known genetic changes account for only a small proportion of cases of idiopathic pulmonary fibrosis. However, mutations in genes known as TERC and TERT have been found in about 15 percent of all cases of familial pulmonary fibrosis and a smaller percentage of cases of sporadic idiopathic pulmonary fibrosis. The TERC and TERT genes provide instructions for making components of an enzyme called telomerase, which maintains structures at the ends of chromosomes known as telomeres. A decrease in telomerase function allows telomeres to become abnormally short as cells divide. The shortened telomeres likely trigger lung cells to stop dividing or to die prematurely. In people with idiopathic pulmonary fibrosis, shorter telomeres are associated with a more severe disease and a quicker decline in lung function. Additional research is needed to confirm how shortened telomeres contribute to the progressive scarring and lung damage characteristic of idiopathic pulmonary fibrosis.
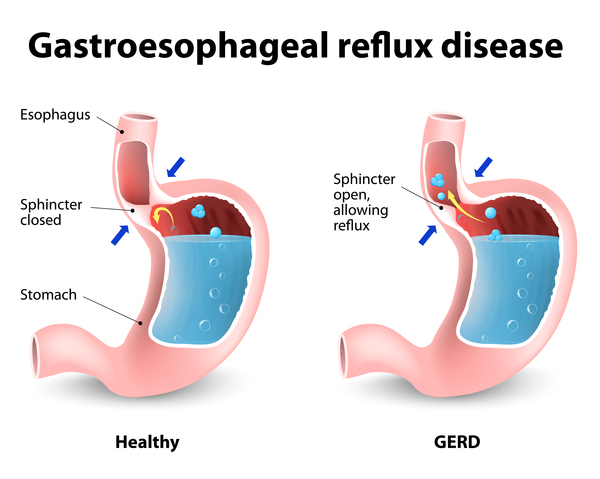
Researchers have also examined environmental risk factors that could contribute to idiopathic pulmonary fibrosis. These factors include exposure to wood or metal dust, viral infections, certain medications, and cigarette smoking. Some research suggests that gastroesophageal reflux disease (GERD) may also be a risk factor for idiopathic pulmonary fibrosis; affected individuals may breathe in (aspirate) stomach contents, which over time could damage the lungs.
Inheritance
Most cases of idiopathic pulmonary fibrosis are sporadic; they occur in people with no history of the disorder in their family.
Familial pulmonary fibrosis appears to have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of an altered gene in each cell is sufficient to cause the disorder. However, some people who inherit the altered gene never develop features of familial pulmonary fibrosis. (This situation is known as reduced penetrance.) It is unclear why some people with a mutated gene develop the disease and other people with the mutated gene do not.

Other Names for This Condition
- Cryptogenic fibrosing alveolitis
- Idiopathic fibrosing alveolitis, chronic form
- IPF
- Usual interstitial pneumonia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, Kraven LM, Obeidat M, Li X, Ng M, Braybrooke R, Molina-Molina M, Hobbs BD, Putman RK, Sakornsakolpat P, Booth HL, Fahy WA, Hart SP, Hill MR, Hirani N, Hubbard RB, McAnulty RJ, Millar AB, Navaratnam V, Oballa E, Parfrey H, Saini G, Whyte MKB, Zhang Y, Kaminski N, Adegunsoye A, Strek ME, Neighbors M, Sheng XR, Gudmundsson G, Gudnason V, Hatabu H, Lederer DJ, Manichaikul A, Newell JD Jr, O'Connor GT, Ortega VE, Xu H, Fingerlin TE, Bosse Y, Hao K, Joubert P, Nickle DC, Sin DD, Timens W, Furniss D, Morris AP, Zondervan KT, Hall IP, Sayers I, Tobin MD, Maher TM, Cho MH, Hunninghake GM, Schwartz DA, Yaspan BL, Molyneaux PL, Flores C, Noth I, Jenkins RG, Wain LV. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2020 Mar 1;201(5):564-574. doi: 10.1164/rccm.201905-1017OC. Citation on PubMed or Free article on PubMed Central
- Allen RJ, Porte J, Braybrooke R, Flores C, Fingerlin TE, Oldham JM, Guillen-Guio B, Ma SF, Okamoto T, John AE, Obeidat M, Yang IV, Henry A, Hubbard RB, Navaratnam V, Saini G, Thompson N, Booth HL, Hart SP, Hill MR, Hirani N, Maher TM, McAnulty RJ, Millar AB, Molyneaux PL, Parfrey H, Rassl DM, Whyte MKB, Fahy WA, Marshall RP, Oballa E, Bosse Y, Nickle DC, Sin DD, Timens W, Shrine N, Sayers I, Hall IP, Noth I, Schwartz DA, Tobin MD, Wain LV, Jenkins RG. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med. 2017 Nov;5(11):869-880. doi: 10.1016/S2213-2600(17)30387-9. Epub 2017 Oct 20. Citation on PubMed or Free article on PubMed Central
- American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000 Feb;161(2 Pt 1):646-64. doi: 10.1164/ajrccm.161.2.ats3-00. No abstract available. Citation on PubMed
- Arish N, Petukhov D, Wallach-Dayan SB. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. Int J Mol Sci. 2019 Jun 19;20(12):2996. doi: 10.3390/ijms20122996. Citation on PubMed or Free article on PubMed Central
- Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007 Mar 29;356(13):1317-26. doi: 10.1056/NEJMoa066157. Citation on PubMed
- Borchers AT, Chang C, Keen CL, Gershwin ME. Idiopathic pulmonary fibrosis-an epidemiological and pathological review. Clin Rev Allergy Immunol. 2011 Apr;40(2):117-34. doi: 10.1007/s12016-010-8211-5. Citation on PubMed
- Garcia CK, Talbert JL. Pulmonary Fibrosis Predisposition Overview. 2005 Jan 21 [updated 2022 May 12]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1230/ Citation on PubMed
- Harari S, Caminati A. IPF: new insight on pathogenesis and treatment. Allergy. 2010 May;65(5):537-53. doi: 10.1111/j.1398-9995.2009.02305.x. Epub 2010 Feb 1. Citation on PubMed
- Hobbs BD, Putman RK, Araki T, Nishino M, Gudmundsson G, Gudnason V, Eiriksdottir G, Zilhao Nogueira NR, Dupuis J, Xu H, O'Connor GT, Manichaikul A, Nguyen J, Podolanczuk AJ, Madahar P, Rotter JI, Lederer DJ, Barr RG, Rich SS, Ampleford EJ, Ortega VE, Peters SP, O'Neal WK, Newell JD Jr, Bleecker ER, Meyers DA, Allen RJ, Oldham JM, Ma SF, Noth I, Jenkins RG, Maher TM, Hubbard RB, Wain LV, Fingerlin TE, Schwartz DA, Washko GR, Rosas IO, Silverman EK, Hatabu H, Cho MH, Hunninghake GM. Overlap of Genetic Risk between Interstitial Lung Abnormalities and Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019 Dec 1;200(11):1402-1413. doi: 10.1164/rccm.201903-0511OC. Citation on PubMed or Free article on PubMed Central
- Kolb M, Vasakova M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res. 2019 Mar 14;20(1):57. doi: 10.1186/s12931-019-1022-1. Citation on PubMed or Free article on PubMed Central
- McLean-Tooke A, Moore I, Lake F. Idiopathic and immune-related pulmonary fibrosis: diagnostic and therapeutic challenges. Clin Transl Immunology. 2019 Nov 5;8(11):e1086. doi: 10.1002/cti2.1086. eCollection 2019. Citation on PubMed or Free article on PubMed Central
- Ryu JH, Moua T, Daniels CE, Hartman TE, Yi ES, Utz JP, Limper AH. Idiopathic pulmonary fibrosis: evolving concepts. Mayo Clin Proc. 2014 Aug;89(8):1130-42. doi: 10.1016/j.mayocp.2014.03.016. Epub 2014 May 24. Citation on PubMed
- Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, Torres F, Kaza V, Girod CE, Jones KD, Elicker BM, Ma SF, Vij R, Collard HR, Wolters PJ, Garcia CK. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. 2014 Jul;2(7):557-65. doi: 10.1016/S2213-2600(14)70124-9. Epub 2014 Jun 16. Citation on PubMed or Free article on PubMed Central
- Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007 May 1;104(18):7552-7. doi: 10.1073/pnas.0701009104. Epub 2007 Apr 25. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.