

Description
As its name suggests, a Huntington's disease-like (HDL) syndrome is a condition that resembles Huntington's disease. Researchers have described four HDL syndromes, designated Huntington's disease-like 1 (HDL1) through Huntington's disease-like 4 (HDL4). These progressive brain disorders are characterized by uncontrolled movements, emotional problems, and loss of thinking ability. HDL syndromes occur in people with the characteristic features of Huntington's disease who do not have a variant (also called mutation) in the gene typically associated with that disorder.
HDL1, HDL2, and HDL4 usually appear in early to mid-adulthood, although they can begin earlier in life. The first signs and symptoms of these conditions often include irritability, emotional problems, small involuntary movements, poor coordination, and trouble learning new information or making decisions. Many affected people develop involuntary jerking or twitching movements known as chorea. As the disease progresses, these abnormal movements become more pronounced. Affected individuals may develop problems with walking, speaking, and swallowing. People with these disorders also experience changes in personality and a decline in thinking and reasoning abilities. Individuals with an HDL syndrome can live for a few years to more than a decade after signs and symptoms begin.
HDL3 begins much earlier in life than most of the other HDL syndromes (usually around age 3 or 4). Affected children experience a decline in thinking ability, difficulties with movement and speech, and seizures. Because HDL3 has a somewhat different pattern of signs and symptoms and a different pattern of inheritance, researchers are unsure whether it belongs in the same category as the other HDL syndromes.
Frequency
Overall, HDL syndromes are rare. They are much less common than Huntington's disease, which affects an estimated 3 to 7 per 100,000 people of European ancestry.
Of the four described HDL syndromes, HDL4 appears to be the most common. HDL2 is the second most common and occurs almost exclusively in people of African heritage (especially Black South Africans). HDL1 has been reported in only one family. HDL3 has been found in two families, both of which were from Saudi Arabia.
Causes
In about one percent of people with the characteristic features of Huntington disease, no variants in the HTT gene has been identified. Variants in the PRNP, JPH3, and TBP genes have been found to cause the signs and symptoms in some of these individuals. HDL1 is caused by variants in the PRNP gene, while HDL2 results from variants in JPH3. Variants in the TBP gene are responsible for HDL4 (also known as spinocerebellar ataxia type 17). The genetic cause of HDL3 is unknown.
The PRNP, JPH3, and TBP genes provide instructions for making proteins that are important for normal brain function. The features of HDL syndromes result from a particular type of variant in any one of these genes. This variant increases the length of a repeated segment of DNA within the gene, which leads to the production of an abnormal PRNP, JPH3, or TBP protein. The abnormal protein can build up in nerve cells (neurons) and disrupt the normal functions of these cells. The dysfunction and eventual death of neurons in certain areas of the brain underlie the signs and symptoms of HDL syndromes.
Other medical conditions and gene variant may also cause signs and symptoms resembling Huntington's disease. In some affected people, the cause of the disorder is never identified.
Inheritance
HDL1, HDL2, and HDL4 are inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.
As the variant responsible for HDL2 or HDL4 is passed down from one generation to the next, the length of the repeated DNA segment may increase. A longer repeat segment is often associated with more severe signs and symptoms that appear earlier in life. This phenomenon is known as anticipation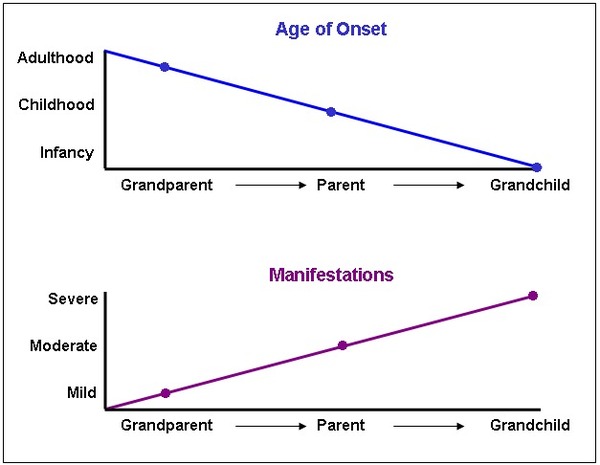 .
.
HDL3 is probably inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Huntington disease-like syndrome
- Huntington disease-like syndromes
- Huntington's disease phenocopies
- Huntington's disease phenocopy syndromes
- Huntington's disease-like syndromes
Additional Information & Resources
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Schneider SA, Walker RH, Bhatia KP. The Huntington's disease-like syndromes: what to consider in patients with a negative Huntington's disease gene test. Nat Clin Pract Neurol. 2007 Sep;3(9):517-25. doi: 10.1038/ncpneuro0606. Citation on PubMed
- Stevanin G, Fujigasaki H, Lebre AS, Camuzat A, Jeannequin C, Dode C, Takahashi J, San C, Bellance R, Brice A, Durr A. Huntington's disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain. 2003 Jul;126(Pt 7):1599-603. doi: 10.1093/brain/awg155. Epub 2003 May 6. Citation on PubMed
- Wild EJ, Mudanohwo EE, Sweeney MG, Schneider SA, Beck J, Bhatia KP, Rossor MN, Davis MB, Tabrizi SJ. Huntington's disease phenocopies are clinically and genetically heterogeneous. Mov Disord. 2008 Apr 15;23(5):716-20. doi: 10.1002/mds.21915. Citation on PubMed
- Wild EJ, Tabrizi SJ. Huntington's disease phenocopy syndromes. Curr Opin Neurol. 2007 Dec;20(6):681-7. doi: 10.1097/WCO.0b013e3282f12074. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.