

Description
Hereditary paraganglioma-pheochromocytoma is an inherited condition characterized by the growth of tumors in structures called paraganglia. Paraganglia are groups of cells that are found near nerve cell bunches called ganglia. A tumor involving the paraganglia is known as a paraganglioma. A type of paraganglioma known as a pheochromocytoma develops in the adrenal glands, which are located on top of each kidney and produce hormones in response to stress. Other types of paraganglioma are usually found in the head, neck, or trunk. People with hereditary paraganglioma-pheochromocytoma develop one or more paragangliomas, which may include pheochromocytomas.
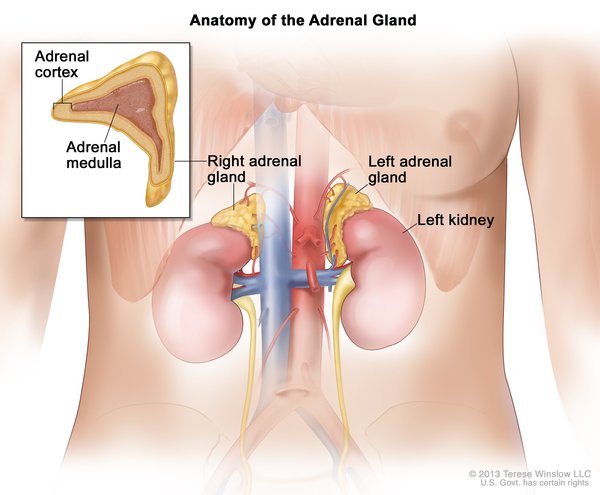
Pheochromocytomas and some other paragangliomas are associated with ganglia of the sympathetic nervous system. The sympathetic nervous system controls the "fight-or-flight" response, a series of changes in the body due to hormones released in response to stress. Sympathetic paragangliomas found outside the adrenal glands, usually in the abdomen, are called extra-adrenal paragangliomas. Most sympathetic paragangliomas, including pheochromocytomas, produce hormones called catecholamines, such as epinephrine (adrenaline) or norepinephrine. These excess catecholamines can cause signs and symptoms such as high blood pressure (hypertension), episodes of rapid heartbeat (palpitations), headaches, or sweating.
Most paragangliomas are associated with ganglia of the parasympathetic nervous system, which controls involuntary body functions such as digestion and saliva formation. Parasympathetic paragangliomas, typically found in the head and neck, usually do not produce hormones. However, large tumors may cause signs and symptoms such as coughing, hearing loss in one ear, or difficulty swallowing.
Paragangliomas and pheochromocytomas are typically considered an undetermined tumor type, meaning they can be noncancerous (benign) or become cancerous (malignant) and spread to other parts of the body (metastasize). Extra-adrenal paragangliomas become malignant more often than other types of paraganglioma or pheochromocytoma.
Researchers have identified several types of hereditary paraganglioma-pheochromocytoma. Each type is distinguished by its genetic cause. People with types 1, 2, and 3 typically develop paragangliomas in the head or neck region. People with type 4 usually develop extra-adrenal paragangliomas in the abdomen and are at higher risk for malignant tumors that metastasize. The other types are very rare. Hereditary paraganglioma-pheochromocytoma is typically diagnosed in a person's 30s.
Paragangliomas and pheochromocytomas can occur in individuals with other inherited disorders, such as von Hippel-Lindau syndrome, Carney-Stratakis syndrome, and certain types of multiple endocrine neoplasia. These other disorders feature additional tumor types and have different genetic causes. Some paragangliomas and pheochromocytomas occur in people with no history of the tumors in their families and appear not to be inherited. These cases are designated as sporadic.
Frequency
Hereditary paraganglioma-pheochromocytoma occurs in approximately 1 in 1 million people.
Causes
Mutations in at least four genes increase the risk of developing the different types of hereditary paraganglioma-pheochromocytoma. Mutations in the SDHD gene predispose an individual to hereditary paraganglioma-pheochromocytoma type 1; mutations in the SDHAF2 gene predispose to type 2; mutations in the SDHC gene predispose to type 3; and mutations in the SDHB gene predispose to type 4.
The SDHB, SDHC, and SDHD genes provide instructions for making three of the four subunits of an enzyme called succinate dehydrogenase (SDH). In addition, the protein made by the SDHAF2 gene is required for the SDH enzyme to function. The SDH enzyme links two important cellular pathways called the citric acid cycle (or Krebs cycle) and oxidative phosphorylation. These pathways are critical in converting the energy from food into a form that cells can use.
As part of the citric acid cycle, the SDH enzyme converts a compound called succinate to another compound called fumarate. Succinate acts as an oxygen sensor in the cell and can help turn on specific pathways that stimulate cells to grow in a low-oxygen environment (hypoxia).
Mutations in the SDHB, SDHC, SDHD, and SDHAF2 genes lead to the loss or reduction of SDH enzyme activity. Because the mutated SDH enzyme cannot convert succinate to fumarate, succinate accumulates in the cell. As a result, the hypoxia pathways are triggered in normal oxygen conditions, which lead to abnormal cell growth and tumor formation.
Inheritance
Hereditary paraganglioma-pheochromocytoma is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to increase the risk of developing tumors. An additional mutation that deletes the normal copy of the gene is needed to cause the condition. This second mutation, called a somatic mutation, is acquired during a person's lifetime and is present only in tumor cells.

The risk of developing hereditary paraganglioma-pheochromocytoma types 1 and 2 is passed on only if the mutated copy of the gene is inherited from the father. The mechanism of this pattern of inheritance is unknown. The risk of developing types 3 and 4 can be inherited from the mother or the father.
Other Names for This Condition
- Familial paraganglioma syndrome
- Familial paraganglioma-pheochromocytoma syndromes
- FPGL
- FPGL/PHEO
- Hereditary paraganglioma-pheochromocytoma syndromes
- Hereditary pheochromocytoma-paraganglioma
- Paragangliomas 1
- Paragangliomas 2
- Paragangliomas 3
- Paragangliomas 4
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001 Jul;69(1):49-54. doi: 10.1086/321282. Epub 2001 Jun 12. Erratum In: Am J Hum Genet 2002 Feb;70(2):565. Citation on PubMed or Free article on PubMed Central
- Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJ, Hoefsloot LH, Hermsen MA, Suarez C, Hussain AK, Vriends AH, Hes FJ, Jansen JC, Tops CM, Corssmit EP, de Knijff P, Lenders JW, Cremers CW, Devilee P, Dinjens WN, de Krijger RR, Robledo M. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010 Apr;11(4):366-72. doi: 10.1016/S1470-2045(10)70007-3. Epub 2010 Jan 11. Citation on PubMed
- Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, Niccoli P, Gaillard D, Chabrier G, Chabolle F, Coupier I, Thieblot P, Lecomte P, Bertherat J, Wion-Barbot N, Murat A, Venisse A, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP; PGL.NET network. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009 Aug;94(8):2817-27. doi: 10.1210/jc.2008-2504. Epub 2009 May 19. Citation on PubMed
- Chetty R. Familial paraganglioma syndromes. J Clin Pathol. 2010 Jun;63(6):488-91. doi: 10.1136/jcp.2010.076257. Citation on PubMed
- Crona J, Taieb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr Rev. 2017 Dec 1;38(6):489-515. doi: 10.1210/er.2017-00062. Citation on PubMed or Free article on PubMed Central
- Douwes Dekker PB, Hogendoorn PC, Kuipers-Dijkshoorn N, Prins FA, van Duinen SG, Taschner PE, van der Mey AG, Cornelisse CJ. SDHD mutations in head and neck paragangliomas result in destabilization of complex II in the mitochondrial respiratory chain with loss of enzymatic activity and abnormal mitochondrial morphology. J Pathol. 2003 Nov;201(3):480-6. doi: 10.1002/path.1461. Citation on PubMed
- Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. 2008 May 21 [updated 2023 Sep 21]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1548/ Citation on PubMed
- Gimenez-Roqueplo AP, Favier J, Rustin P, Mourad JJ, Plouin PF, Corvol P, Rotig A, Jeunemaitre X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001 Dec;69(6):1186-97. doi: 10.1086/324413. Epub 2001 Oct 16. Citation on PubMed or Free article on PubMed Central
- Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X; COMETE Network. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003 Sep 1;63(17):5615-21. Citation on PubMed
- Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009 Aug 28;325(5944):1139-42. doi: 10.1126/science.1175689. Epub 2009 Jul 23. Citation on PubMed or Free article on PubMed Central
- Muller U. Pathological mechanisms and parent-of-origin effects in hereditary paraganglioma/pheochromocytoma (PGL/PCC). Neurogenetics. 2011 Aug;12(3):175-81. doi: 10.1007/s10048-011-0280-y. Epub 2011 Mar 9. Citation on PubMed
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C; European-American Paraganglioma Study Group. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004 Aug 25;292(8):943-51. doi: 10.1001/jama.292.8.943. Erratum In: JAMA. 2004 Oct 13;292(14):1686. Citation on PubMed
- Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000 Nov;26(3):268-70. doi: 10.1038/81551. Citation on PubMed
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med. 2009 Jul;266(1):19-42. doi: 10.1111/j.1365-2796.2009.02111.x. Citation on PubMed or Free article on PubMed Central
- Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005 Aug 1;14(15):2231-9. doi: 10.1093/hmg/ddi227. Epub 2005 Jun 29. Citation on PubMed
- Schiavi F, Boedeker CC, Bausch B, Peczkowska M, Gomez CF, Strassburg T, Pawlu C, Buchta M, Salzmann M, Hoffmann MM, Berlis A, Brink I, Cybulla M, Muresan M, Walter MA, Forrer F, Valimaki M, Kawecki A, Szutkowski Z, Schipper J, Walz MK, Pigny P, Bauters C, Willet-Brozick JE, Baysal BE, Januszewicz A, Eng C, Opocher G, Neumann HP; European-American Paraganglioma Study Group. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005 Oct 26;294(16):2057-63. doi: 10.1001/jama.294.16.2057. Erratum In: JAMA. 2006 Feb 8;295(6):628. Citation on PubMed
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005 Jan;7(1):77-85. doi: 10.1016/j.ccr.2004.11.022. Citation on PubMed
- Williams MD, Tischler AS. Update from the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Paragangliomas. Head Neck Pathol. 2017 Mar;11(1):88-95. doi: 10.1007/s12105-017-0786-1. Epub 2017 Feb 28. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.