

Description
Hereditary hypophosphatemic rickets is a disorder related to low levels of phosphate in the blood (hypophosphatemia). Phosphate is a mineral that is essential for the normal formation of bones and teeth.
In most cases, the signs and symptoms of hereditary hypophosphatemic rickets begin in early childhood. The features of the disorder vary widely, even among affected members of the same family. Mildly affected individuals may have hypophosphatemia without other signs and symptoms. More severely affected children experience slow growth and are shorter than their peers. They develop bone abnormalities that can interfere with movement and cause bone pain. The most noticeable of these abnormalities are bowed legs or knock knees. These abnormalities become apparent with weight-bearing activities such as walking. If untreated, they tend to worsen with time.
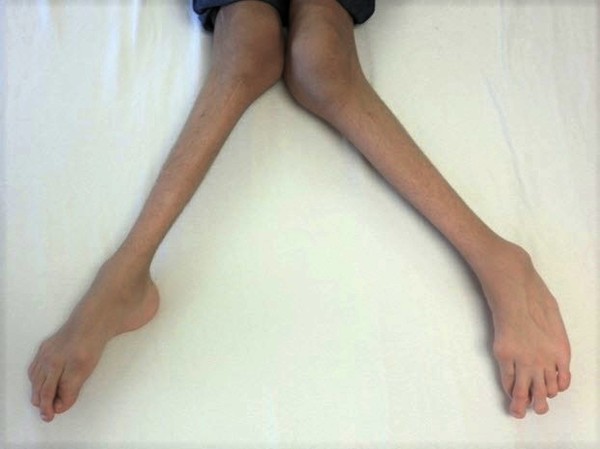
Other signs and symptoms of hereditary hypophosphatemic rickets can include premature fusion of the skull bones (craniosynostosis) and dental abnormalities. The disorder may also cause abnormal bone growth where ligaments and tendons attach to joints (enthesopathy). In adults, hypophosphatemia is characterized by a softening of the bones known as osteomalacia.
Researchers have described several forms of hereditary hypophosphatemic rickets, which are distinguished by their pattern of inheritance and genetic cause. The most common form of the disorder is known as X-linked hypophosphatemic rickets (XLH). It has an X-linked dominant pattern of inheritance. X-linked recessive, autosomal dominant, and autosomal recessive forms of the disorder are much rarer.
Another rare type of the disorder is known as hereditary hypophosphatemic rickets with hypercalciuria (HHRH). In addition to hypophosphatemia, this condition is characterized by the excretion of high levels of calcium in the urine (hypercalciuria).
Frequency
X-linked hypophosphatemic rickets is the most common form of rickets that runs in families. It affects about 1 in 20,000 newborns. Each of the other forms of hereditary hypophosphatemic rickets has been identified in only a few families.
Causes
Hereditary hypophosphatemic rickets can result from mutations in several genes. Mutations in the PHEX gene, which are responsible for X-linked hypophosphatemic rickets, occur most frequently. Mutations in other genes cause the less common forms of the condition.
Hereditary hypophosphatemic rickets is characterized by a phosphate imbalance in the body. Among its many functions, phosphate plays a critical role in the formation and growth of bones in childhood and helps maintain bone strength in adults. Phosphate levels are controlled in large part by the kidneys. The kidneys normally excrete excess phosphate in urine, and they reabsorb this mineral into the bloodstream when more is needed. However, in people with hereditary hypophosphatemic rickets, the kidneys cannot reabsorb phosphate effectively and too much of this mineral is excreted from the body in urine. As a result, not enough phosphate is available in the bloodstream to participate in normal bone development and maintenance.
The genes associated with hereditary hypophosphatemic rickets are involved in maintaining the proper balance of phosphate. Many of these genes, including the PHEX gene, directly or indirectly regulate a protein called fibroblast growth factor 23 (produced from the FGF23 gene). This protein normally inhibits the kidneys' ability to reabsorb phosphate into the bloodstream. Gene mutations increase the production or reduce the breakdown of fibroblast growth factor 23. The resulting overactivity of this protein reduces phosphate reabsorption by the kidneys, leading to hypophosphatemia and the related features of hereditary hypophosphatemic rickets.
Inheritance
Hereditary hypophosphatemic rickets can have several patterns of inheritance. When the condition results from mutations in the PHEX gene, it is inherited in an X-linked dominant pattern. The PHEX gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
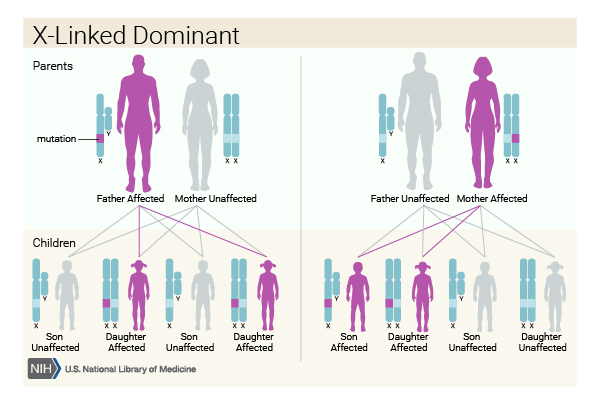

Less commonly, hereditary hypophosphatemic rickets can have an X-linked recessive pattern of inheritance. This form of the condition is often called Dent disease. Like the PHEX gene, the gene associated with Dent disease is located on the X chromosome. In males, one altered copy of the gene in each cell is sufficient to cause the condition. In females, a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females.

In a few families, hereditary hypophosphatemic rickets has had an autosomal dominant inheritance pattern, which means one copy of an altered gene in each cell is sufficient to cause the disorder. The rare condition HHRH has an autosomal recessive pattern of inheritance, which means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. However, some parents of children with HHRH have experienced hypercalcuria and kidney stones.


Other Names for This Condition
- Hypophosphatemia
- VDRR
- Vitamin D-resistant rickets
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Autosomal dominant hypophosphatemic rickets

- Genetic Testing Registry: Autosomal recessive hypophosphatemic bone disease
- Genetic Testing Registry: Familial X-linked hypophosphatemic vitamin D refractory rickets
- Genetic Testing Registry: Hypophosphatemic rickets, autosomal recessive, 1
- Genetic Testing Registry: Hypophosphatemic rickets, autosomal recessive, 2
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- HYPOPHOSPHATEMIC RICKETS, AUTOSOMAL DOMINANT; ADHR
- HYPOPHOSPHATEMIC RICKETS, X-LINKED DOMINANT; XLHR
- HYPOPHOSPHATEMIC RICKETS, X-LINKED RECESSIVE
- HYPOPHOSPHATEMIC RICKETS, AUTOSOMAL RECESSIVE, 1; ARHR1
- HYPOPHOSPHATEMIC RICKETS WITH HYPERCALCIURIA, HEREDITARY; HHRH
- HYPOPHOSPHATEMIC RICKETS, AUTOSOMAL RECESSIVE, 2; ARHR2
Scientific Articles on PubMed
References
- Baroncelli GI, Bertelloni S, Sodini F, Galli L, Vanacore T, Fiore L, Saggese G. Genetic advances, biochemical and clinical features and critical approach to treatment of patients with X-linked hypophosphatemic rickets. Pediatr Endocrinol Rev. 2004 Jun;1(4):361-79. Citation on PubMed
- DiMeglio LA, Econs MJ. Hypophosphatemic rickets. Rev Endocr Metab Disord. 2001 Apr;2(2):165-73. doi: 10.1023/a:1010054727323. No abstract available. Citation on PubMed
- Nield LS, Mahajan P, Joshi A, Kamat D. Rickets: not a disease of the past. Am Fam Physician. 2006 Aug 15;74(4):619-26. Citation on PubMed
- Pettifor JM. What's new in hypophosphataemic rickets? Eur J Pediatr. 2008 May;167(5):493-9. doi: 10.1007/s00431-007-0662-1. Epub 2008 Jan 24. Citation on PubMed or Free article on PubMed Central
- Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab. 2009;27(4):392-401. doi: 10.1007/s00774-009-0079-1. Epub 2009 Jun 6. Citation on PubMed
- Wharton B, Bishop N. Rickets. Lancet. 2003 Oct 25;362(9393):1389-400. doi: 10.1016/S0140-6736(03)14636-3. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.