

Description
Glanzmann thrombasthenia is a bleeding disorder that is characterized by prolonged or spontaneous bleeding starting from birth. People with Glanzmann thrombasthenia tend to bruise easily, have frequent nosebleeds (epistaxis), and may bleed from the gums. They may also develop red or purple spots on the skin caused by bleeding underneath the skin (petechiae) or swelling caused by bleeding within tissues (hematoma). Glanzmann thrombasthenia can also cause prolonged bleeding following injury, trauma, or surgery (including dental work). Women with this condition can have prolonged and sometimes abnormally heavy menstrual bleeding. Affected women also have an increased risk of excessive blood loss during pregnancy and childbirth.
About a quarter of individuals with Glanzmann thrombasthenia have bleeding in the gastrointestinal tract, which often occurs later in life. Rarely, affected individuals have bleeding inside the skull (intracranial hemorrhage) or joints (hemarthrosis).
The severity and frequency of the bleeding episodes in Glanzmann thrombasthenia can vary greatly among affected individuals, even in the same family. Spontaneous bleeding tends to become less frequent with age.
Frequency
Glanzmann thrombasthenia is estimated to affect 1 in one million individuals worldwide, but may be more common in certain groups, including those of Romani ethnicity, particularly people within the French Manouche community.
Causes
Mutations in the ITGA2B or ITGB3 gene cause Glanzmann thrombasthenia. These genes provide instructions for making the two parts (subunits) of a receptor protein called integrin alphaIIb/beta3 (αIIbβ3). This protein is abundant on the surface of platelets. Platelets are small cells that circulate in blood and are an essential component of blood clots. During clot formation, integrin αIIbβ3 helps platelets bind together. Blood clots protect the body after injury by sealing off damaged blood vessels and preventing further blood loss.
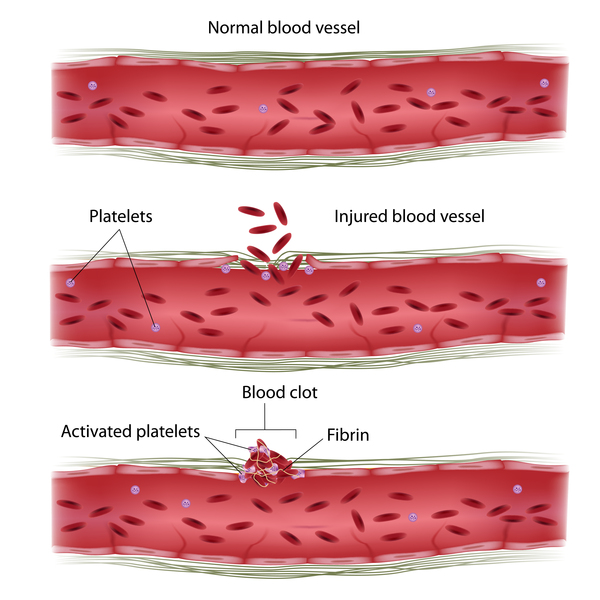
ITGA2B or ITGB3 gene mutations result in a shortage (deficiency) of functional integrin αIIbβ3. As a result, platelets cannot clump together to form a blood clot, leading to prolonged bleeding.
Three types of Glanzmann thrombasthenia have been classified according to the amount of integrin αIIbβ3 that is available. People with type I (the most common type) have less than 5 percent of normal integrin αIIbβ3 levels, people with type II have between 5 and 20 percent of normal integrin αIIbβ3 levels, and people with the variant type have adequate integrin αIIbβ3 levels but produce only nonfunctional integrin.
Some people with Glanzmann thrombasthenia do not have an identified mutation in either the ITGA2B or ITGB3 gene; the cause of the disorder in these individuals is unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Deficiency of glycoprotein complex IIb-IIIa
- Deficiency of platelet fibrinogen receptor
- Glanzmann disease
- Glanzmann-Naegeli disorder
- Glycoprotein IIb/IIIa defect
- Hereditary hemorrhagic thrombasthenia
- Hereditary thrombasthenia
- Platelet fibrinogen receptor deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011 Dec 1;118(23):5996-6005. doi: 10.1182/blood-2011-07-365635. Epub 2011 Sep 13. Citation on PubMed
- Nurden AT, Pillois X, Fiore M, Alessi MC, Bonduel M, Dreyfus M, Goudemand J, Gruel Y, Benabdallah-Guerida S, Latger-Cannard V, Negrier C, Nugent D, Oiron RD, Rand ML, Sie P, Trossaert M, Alberio L, Martins N, Sirvain-Trukniewicz P, Couloux A, Canault M, Fronthroth JP, Fretigny M, Nurden P, Heilig R, Vinciguerra C. Expanding the Mutation Spectrum Affecting alphaIIbbeta3 Integrin in Glanzmann Thrombasthenia: Screening of the ITGA2B and ITGB3 Genes in a Large International Cohort. Hum Mutat. 2015 May;36(5):548-61. doi: 10.1002/humu.22776. Citation on PubMed
- Nurden AT, Pillois X, Wilcox DA. Glanzmann thrombasthenia: state of the art and future directions. Semin Thromb Hemost. 2013 Sep;39(6):642-55. doi: 10.1055/s-0033-1353393. Epub 2013 Aug 8. Citation on PubMed or Free article on PubMed Central
- Pillitteri D, Pilgrimm AK, Kirchmaier CM. Novel Mutations in the GPIIb and GPIIIa Genes in Glanzmann Thrombasthenia. Transfus Med Hemother. 2010;37(5):268-277. doi: 10.1159/000320258. Epub 2010 Sep 15. Citation on PubMed or Free article on PubMed Central
- Sandrock-Lang K, Oldenburg J, Wiegering V, Halimeh S, Santoso S, Kurnik K, Fischer L, Tsakiris DA, Sigl-Kraetzig M, Brand B, Buhrlen M, Kraetzer K, Deeg N, Hund M, Busse E, Kahle A, Zieger B. Characterisation of patients with Glanzmann thrombasthenia and identification of 17 novel mutations. Thromb Haemost. 2015 Apr;113(4):782-91. doi: 10.1160/TH14-05-0479. Epub 2014 Nov 6. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.