

Description
Genetic epilepsy with febrile seizures plus (GEFS+) is a spectrum of seizure disorders of varying severity. GEFS+ is usually diagnosed in families whose members have a combination of febrile seizures, which are triggered by a high fever, and recurrent seizures (epilepsy) of other types, including seizures that are not related to fevers (afebrile seizures). The additional seizure types usually involve both sides of the brain (generalized seizures); however, seizures that involve only one side of the brain (partial seizures) occur in some affected individuals. The most common types of seizure in people with GEFS+ include myoclonic seizures, which cause involuntary muscle twitches; atonic seizures, which involve sudden episodes of weak muscle tone; and absence seizures, which cause loss of consciousness for short periods that appear as staring spells.
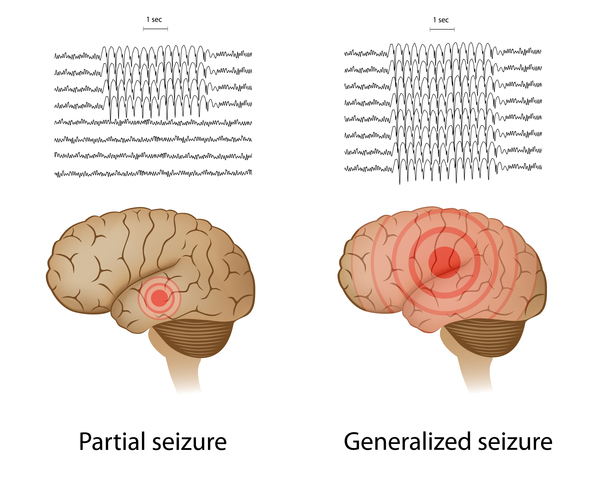
The most common and mildest feature of the GEFS+ spectrum is simple febrile seizures, which begin in infancy and usually stop by age 5. When the febrile seizures continue after age 5 or other types of seizure develop, the condition is called febrile seizures plus (FS+). Seizures in FS+ usually end in early adolescence.
A condition called Dravet syndrome (also known as severe myoclonic epilepsy of infancy or SMEI) is often considered part of the GEFS+ spectrum and is the most severe disorder in this group. Affected infants typically have prolonged seizures lasting several minutes (status epilepticus), which are triggered by fever. Other seizure types, including afebrile seizures, begin in early childhood. These types can include myoclonic or absence seizures. In Dravet syndrome, these seizures are difficult to control with medication, and they can worsen over time. A decline in brain function is also common in Dravet syndrome. Affected individuals usually develop normally in the first year of life, but then development stalls, and some affected children lose already-acquired skills (developmental regression). Many people with Dravet syndrome have difficulty coordinating movements (ataxia) and intellectual disability.
Some people with GEFS+ have seizure disorders of intermediate severity that may not fit into the classical diagnosis of simple febrile seizures, FS+, or Dravet syndrome.
Family members with GEFS+ may have different combinations of febrile seizures and epilepsy. For example, one affected family member may have only febrile seizures, while another also has myoclonic epilepsy. While GEFS+ is usually diagnosed in families, it can occur in individuals with no history of the condition in their family.
Frequency
GEFS+ is a rare condition. Its prevalence is unknown.
Causes
Mutations in several genes, including some that have not been identified, can cause GEFS+. The most commonly associated gene is SCN1A. More than 80 percent of Dravet syndrome cases and about 10 percent of other GEFS+ cases are caused by changes in this gene. Mutations in other genes have been found in only a small number of affected individuals or families.
The SCN1A gene and others associated with GEFS+ provide instructions for making pieces (subunits) of channels that transport positively charged sodium atoms (sodium ions) into cells. The transport of these ions helps generate and transmit electrical signals between nerve cells (neurons). Proteins produced from other genes involved in GEFS+ are subunits of another type of ion channel called the GABAA receptor. GABAA receptor channels block (inhibit) signaling between neurons. Still other GEFS+-associated genes are also involved in nerve signaling.
Mutations in the SCN1A gene have a variety of effects on sodium channels. Many mutations that cause Dravet syndrome reduce the number of functional channels in each cell. Mutations that cause the milder GEFS+ disorders likely alter the channel's structure. All of these genetic changes affect the ability of the channels to transport sodium ions into neurons. Some mutations are thought to reduce channel activity while others may increase it. It is unclear, however, how these changes underlie the development of seizures. Some studies show that certain SCN1A gene mutations cause signaling between neurons to be constantly turned on (stimulated). Researchers believe that overstimulation of certain neurons in the brain triggers the abnormal brain activity associated with seizures. It is not known if all SCN1A gene mutations have the same effect.
Changes in GABAA receptor subunit genes impair the channel's function, causing uncontrolled signaling between neurons, which likely leads to seizures.
Researchers do not understand how changes in any one of the genes associated with GEFS+ can lead to a range of seizure disorders. Because the disorders are so varied, even among family members, researchers believe that other genes and environmental factors help determine the severity of the condition.
Inheritance
GEFS+ is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new (de novo) mutations in the gene and occur in people with no history of the disorder in their family.


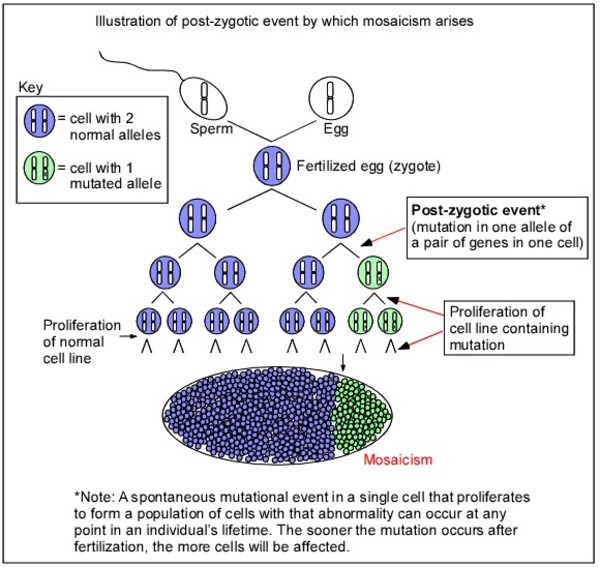
Dravet syndrome is almost always caused by de novo mutations, although it can be inherited from a parent who has a milder form of GEFS+. Other forms of GEFS+ are usually inherited from an affected parent. Rarely, Dravet syndrome or other forms of GEFS+ are inherited from a parent with somatic mosaicism. Somatic mosaicism means that some of the body's cells have the gene mutation, and others do not. A parent with mosaicism may be less severely affected or not show any signs or symptoms of GEFS+.
Other Names for This Condition
- GEFS+
- Generalized epilepsy with febrile seizures plus
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Generalized epilepsy with febrile seizures plus

- Genetic Testing Registry: Generalized epilepsy with febrile seizures plus 3
- Genetic Testing Registry: Generalized epilepsy with febrile seizures plus, type 1
- Genetic Testing Registry: Generalized epilepsy with febrile seizures plus, type 2
- Genetic Testing Registry: Generalized epilepsy with febrile seizures plus, type 9
- Genetic Testing Registry: SCN2A-related generalized epilepsy with febrile seizures plus
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
- GENERALIZED EPILEPSY WITH FEBRILE SEIZURES PLUS, TYPE 2; GEFSP2
- GENERALIZED EPILEPSY WITH FEBRILE SEIZURES PLUS, TYPE 1; GEFSP1
- FEBRILE SEIZURES, FAMILIAL, 8; FEB8
- GENERALIZED EPILEPSY WITH FEBRILE SEIZURES PLUS, TYPE 9; GEFSP9
- EPILEPSY, IDIOPATHIC GENERALIZED, SUSCEPTIBILITY TO, 10; EIG10
- GENERALIZED EPILEPSY WITH FEBRILE SEIZURES PLUS, TYPE 7; GEFSP7
Scientific Articles on PubMed
References
- Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, Berkovic SF, Macdonald RL, Mulley JC. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004 Jul 1;13(13):1315-9. doi: 10.1093/hmg/ddh146. Epub 2004 Apr 28. Citation on PubMed
- Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010 Jun 1;588(Pt 11):1861-9. doi: 10.1113/jphysiol.2010.186999. Epub 2010 Mar 22. Citation on PubMed or Free article on PubMed Central
- Martin MS, Dutt K, Papale LA, Dube CM, Dutton SB, de Haan G, Shankar A, Tufik S, Meisler MH, Baram TZ, Goldin AL, Escayg A. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. 2010 Mar 26;285(13):9823-9834. doi: 10.1074/jbc.M109.078568. Epub 2010 Jan 25. Citation on PubMed or Free article on PubMed Central
- Myers KA, Burgess R, Afawi Z, Damiano JA, Berkovic SF, Hildebrand MS, Scheffer IE. De novo SCN1A pathogenic variants in the GEFS+ spectrum: Not always a familial syndrome. Epilepsia. 2017 Feb;58(2):e26-e30. doi: 10.1111/epi.13649. Epub 2017 Jan 13. Citation on PubMed
- Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev. 2009 May;31(5):394-400. doi: 10.1016/j.braindev.2009.01.001. Epub 2009 Feb 8. Citation on PubMed
- Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, Muhle H, Suls A, Lemke JR, de Kovel CG, Thiele H, Konrad K, Kawalia A, Toliat MR, Sander T, Ruschendorf F, Caliebe A, Nagel I, Kohl B, Kecskes A, Jacmin M, Hardies K, Weckhuysen S, Riesch E, Dorn T, Brilstra EH, Baulac S, Moller RS, Hjalgrim H, Koeleman BP; EuroEPINOMICS RES Consortium; Jurkat-Rott K, Lehman-Horn F, Roach JC, Glusman G, Hood L, Galas DJ, Martin B, de Witte PA, Biskup S, De Jonghe P, Helbig I, Balling R, Nurnberg P, Crawford AD, Esguerra CV, Weber YG, Lerche H. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet. 2014 Dec;46(12):1327-32. doi: 10.1038/ng.3130. Epub 2014 Nov 2. Citation on PubMed
- Shi X, Yasumoto S, Kurahashi H, Nakagawa E, Fukasawa T, Uchiya S, Hirose S. Clinical spectrum of SCN2A mutations. Brain Dev. 2012 Aug;34(7):541-5. doi: 10.1016/j.braindev.2011.09.016. Epub 2011 Oct 24. Citation on PubMed
- Shi YW, Yu MJ, Long YS, Qin B, He N, Meng H, Liu XR, Deng WY, Gao MM, Yi YH, Li BM, Liao WP. Mosaic SCN1A mutations in familial partial epilepsy with antecedent febrile seizures. Genes Brain Behav. 2012 Mar;11(2):170-6. doi: 10.1111/j.1601-183X.2011.00756.x. Epub 2011 Dec 14. Citation on PubMed
- Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, Thompson J, Dixon M, Gurnett C, Peiffer A, White HS, Filloux F, Leppert MF. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009 Sep;5(9):e1000649. doi: 10.1371/journal.pgen.1000649. Epub 2009 Sep 18. Citation on PubMed or Free article on PubMed Central
- Spampanato J, Escayg A, Meisler MH, Goldin AL. Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci. 2001 Oct 1;21(19):7481-90. doi: 10.1523/JNEUROSCI.21-19-07481.2001. Citation on PubMed
- Volkers L, Kahlig KM, Verbeek NE, Das JH, van Kempen MJ, Stroink H, Augustijn P, van Nieuwenhuizen O, Lindhout D, George AL Jr, Koeleman BP, Rook MB. Nav 1.1 dysfunction in genetic epilepsy with febrile seizures-plus or Dravet syndrome. Eur J Neurosci. 2011 Oct;34(8):1268-75. doi: 10.1111/j.1460-9568.2011.07826.x. Epub 2011 Aug 22. Citation on PubMed or Free article on PubMed Central
- Wallace RH, Scheffer IE, Parasivam G, Barnett S, Wallace GB, Sutherland GR, Berkovic SF, Mulley JC. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology. 2002 May 14;58(9):1426-9. doi: 10.1212/wnl.58.9.1426. Citation on PubMed
- Zou F, McWalter K, Schmidt L, Decker A, Picker JD, Lincoln S, Sweetser DA, Briere LC, Harini C; Members of the Undiagnosed Diseases Network; Marsh E, Medne L, Wang RY, Leydiker K, Mower A, Visser G, Cuppen I, van Gassen KL, van der Smagt J, Yousaf A, Tennison M, Shanmugham A, Butler E, Richard G, McKnight D. Expanding the phenotypic spectrum of GABRG2 variants: a recurrent GABRG2 missense variant associated with a severe phenotype. J Neurogenet. 2017 Mar-Jun;31(1-2):30-36. doi: 10.1080/01677063.2017.1315417. Epub 2017 May 2. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.